
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
School of Pharmaceutical Sciences, Starex University.
Objective: The present study was aimed to formulate and evaluate gastroretentive floating beads of Ramipril using a novel foaming technique to prolong gastric residence time and improve bioavailability.Methods: Floating beads were prepared by ionotropic gelation method using sodium alginate as the polymer and Poloxamer 188 as the foaming agent. Five different formulations (F1-F5) were developed by varying sodium alginate concentrations (0.5-2.5% w/v) while keeping Poloxamer 188 (300 mg), CaCl2 (1% w/v), and stirring speed (2600 RPM) constant. Results: The pre-formulation studies confirmed the identity & purity of Ramipril with melting point at 109.6°C & log P value of 3.227. The calibration curve showed linearity (R²=0.994) in the concentration range of 10-100 µg/mL. Among all formulations, F3 (1.5% sodium alginate) exhibited the highest drug entrapment (73.69%) & % yield (72.52%). All formulations showed excellent floating properties with 100% buoyancy for up to 7 hours. The in-vitro drug release from F3 formulation was 93.71% at 24 hours. Release kinetics followed Higuchi model (R²=0.942) with non-Fickian diffusion mechanism (n=0.829).Conclusion: The foaming technique using Poloxamer 188 successfully produced porous floating beads with excellent buoyancy & sustained drug release. The optimized formulation F3 showed promising gastroretentive properties for improved oral delivery of Ramipril.
Cardiovascular diseases (CVDs) remain the leading cause of mortality worldwide, with hypertension being a major pathophysiological trigger for myocardial infarction, stroke, and heart failure [1]. Ramipril, a potent angiotensin-converting enzyme (ACE) inhibitor, is widely prescribed as first-line therapy for hypertension and post-myocardial infarction management. However, its clinical efficacy is limited by poor oral bioavailability (28-35%), short elimination half-life, and dose-dependent adverse effects due to conventional immediate-release formulations [2].
Gastroretentive drug delivery systems (GRDDS) have emerged as promising strategies to overcome these limitations by prolonging gastric residence time and providing sustained drug release [3]. Among various approaches, floating drug delivery systems (FDDS) have gained significant attention due to their ability to remain buoyant in the stomach without affecting gastric emptying [4].
The foaming technique represents a novel approach for fabricating porous floating microspheres. This method introduces an internal gas-generating phase using foaming agents like Poloxamer 188, creating gas-microcavities that lower formulation density below that of gastric fluid (<1.004 g/cm³) [5]. Poloxamer 188, a non-ionic amphiphilic surfactant, effectively reduces surface tension and stabilizes foam through surfactant-polymer complex formation with sodium alginate [6].
The present study was undertaken to develop and evaluate gastroretentive floating beads of Ramipril using Poloxamer 188 as a foaming agent and sodium alginate as a polymer, with the aim of improving bioavailability and patient compliance through sustained drug delivery.
2. MATERIALS AND METHODS
2.1 Materials
Ramipril was obtained as a gift sample. Sodium alginate, Poloxamer 188, calcium chloride, and all other chemicals and reagents used were of analytical grade.
2.2 Pre-formulation Studies
2.2.1 Organoleptic Properties and Melting Point Determination
The drug was evaluated for color, odor, and crystalline nature. Melting point was determined using the capillary fusion method and compared with the reported standard value (106-110°C).
2.2.2 Preparation of Standard Calibration Curve
A stock solution of Ramipril (100 µg/mL) was prepared in methanol. Working standard solutions in the concentration range of 10-100 µg/mL were prepared by serial dilution. The absorbance was measured using a UV-Visible spectrophotometer at λmax 210 nm. The calibration curve was constructed by plotting concentration versus absorbance.
2.2.3 Solubility Studies
The solubility of Ramipril was determined in various solvents including 0.1 N HCl, phosphate buffer pH 6.8, phosphate buffer pH 7.4, and methanol. An excess amount of drug was added to 10 mL of each solvent in sealed vials and shaken for 24 hours at room temperature. The samples were filtered, diluted appropriately, and analyzed spectrophotometrically.
2.2.4 Partition Coefficient Determination
The partition coefficient (log P) was determined using n-octanol and water as the solvent system. An equal volume of both solvents was equilibrated, and the drug was added to the mixture. After shaking for 24 hours, the concentration of drug in both phases was determined and log P was calculated.
2.2.5 FT-IR Spectroscopy
FT-IR spectra of pure Ramipril and the physical mixture of drug and polymers were recorded using the KBr disc method in the range of 4000-400 cm⁻¹.
2.3 Optimization of Foaming Agent
The foamability and foam stability of Poloxamer 188 were evaluated at different concentrations (100-500 mg). Foamability volume was measured after vigorous shaking for 2 minutes, and foam stability was determined by measuring the time required for foam to collapse.
2.4 Preparation of Floating Beads
Floating beads were prepared using the ionotropic gelation method with foam solution. Poloxamer 188 (300 mg) was dissolved in distilled water, and sodium alginate was added at varying concentrations (0.5-2.5% w/v) to prepare foam solutions. The drug was dispersed in the foam solution, and the mixture was stirred at 2600 RPM. The resulting foam solution was extruded through a syringe into 1% calcium chloride solution under continuous stirring. The formed beads were collected, washed, and dried.
Table 1: Batch Specifications of Prepared Floating Beads
|
Batch Code |
Sodium Alginate (%) |
Poloxamer 188 (mg) |
CaCl₂ (%) |
Stirring Speed (RPM) |
|
F1 |
0.5 |
300 |
1 |
2600 |
|
F2 |
1.0 |
300 |
1 |
2600 |
|
F3 |
1.5 |
300 |
1 |
2600 |
|
F4 |
2.0 |
300 |
1 |
2600 |
|
F5 |
2.5 |
300 |
1 |
2600 |
2.5 Evaluation of Floating Beads
2.5.1 Percentage Yield
The percentage yield was calculated as the weight of recovered beads divided by the total weight of drug and polymer used, multiplied by 100.
2.5.2 Drug Entrapment Efficiency
Accurately weighed beads (50 mg) were crushed and dissolved in phosphate buffer pH 7.4. The solution was filtered, and the drug content was determined spectrophotometrically at 210 nm. Drug entrapment was calculated using the formula:
% Drug Entrapment = (Actual drug content / Theoretical drug content) × 100
2.5.3 In-vitro Floating Study
Floating behavior was evaluated in simulated gastric fluid (SGF, pH 1.2). Beads were placed in a beaker containing SGF, and the percentage of floating beads was observed at predetermined time intervals (0-7 hours). The floating lag time was also recorded.
2.5.4 In-vitro Drug Release Study
The in-vitro drug release study was performed using a USP dissolution apparatus (paddle method) at 37±0.5°C in 900 mL of phosphate buffer pH 7.4 at 50 RPM. A mesh was used to ensure complete exposure of the beads. Samples were withdrawn at predetermined time intervals, and the drug content was analyzed spectrophotometrically at 210 nm.
2.6 Drug Release Kinetics
The release data were fitted to various kinetic models including zero-order, first-order, Higuchi, and Korsmeyer-Peppas models. The best-fit model was determined based on the highest regression coefficient (R²) value.
3. RESULTS AND DISCUSSION
3.1 Pre-formulation Studies
3.1.1 Organoleptic Properties and Melting Point
The organoleptic evaluation revealed that Ramipril is a white, odorless, crystalline powder. The melting point was determined to be 109.6°C, which falls within the reported standard range of 106-110°C, confirming the identity and purity of the drug (Table 2).
Table 2: Physicochemical Characterization of Ramipril
|
Parameter |
Observation |
|
Color |
White powder |
|
Odor |
Odorless |
|
Nature |
Crystalline powder |
|
Melting Point |
109.6°C |
|
Loss on Drying |
2.5% w/w |
3.1.2 Standard Calibration Curve
The calibration curve of Ramipril in methanol showed linearity in the concentration range of 10-100 µg/mL at λmax 210 nm. The regression equation was Y = 0.0079x - 0.027 with R² = 0.994, indicating good linearity.
3.1.3 Solubility Studies
The solubility of Ramipril was found to be highly pH-dependent (Table 3). The drug showed the lowest solubility in 0.1 N HCl (0.05 mg/mL) and highest in methanol (1.07 mg/mL). The solubility increased significantly in phosphate buffer pH 7.4 (0.985 mg/mL) compared to acidic medium, indicating that Ramipril is weakly acidic and exists in a less soluble, non-ionized form at gastric pH.
Table 3: Solubility Profile of Ramipril in Different Solvents
|
Solvent |
Solubility (mg/mL) (Mean ± SD) |
|
0.1 N HCl |
0.05 ± 0.0012 |
|
Phosphate Buffer pH 6.8 |
0.267 ± 0.0032 |
|
Phosphate Buffer pH 7.4 |
0.985 ± 0.001 |
|
Methanol |
1.07 ± 0.005 |
3.1.4 Partition Coefficient
The partition coefficient of Ramipril in n-octanol:water was found to be 3.227 ± 0.021. This high log P value indicates that Ramipril is lipophilic in nature, which correlates with its poor aqueous solubility.
3.1.5 FT-IR Spectroscopy
FT-IR spectra of pure Ramipril showed characteristic peaks at 3372 cm⁻¹ (N-H stretch), 2950 cm⁻¹ (C-H stretch), 1682 cm⁻¹ (C=O stretch), 1645 cm⁻¹ (C-N stretch), and 1254 cm⁻¹ (C=C stretch) (Table 6, Figure 3). The FT-IR spectrum of the physical mixture of drug and polymers showed no significant shift in characteristic peaks, indicating no chemical interaction between the drug and polymers (Table 4).
Table 4: Interpretation of FT-IR Spectra of Ramipril
|
Stretching Type |
Standard Range (cm⁻¹) |
Observed Peak (cm⁻¹) |
|
N-H |
3500-3300 |
3372 |
|
C-H |
3000-2850 |
2950 |
|
C=O |
1790-1610 |
1682 |
|
C-N |
1810-1600 |
1645 |
|
C=C |
1350-1210 |
1254 |
3.2 Optimization of Foaming Agent
The foamability and foam stability of Poloxamer 188 were evaluated at different concentrations (100-500 mg) (Table 5). The foamability volume increased from 37.8 mL at 100 mg to a maximum of 53.2 mL at 300 mg, followed by a decrease at higher concentrations. Similarly, foam stability was maximum at 300 mg (12.5 minutes). Therefore, 300 mg of Poloxamer 188 was selected as the optimized concentration for further formulation development.
Table 5: Foamability and Foam Stability of Poloxamer 188
|
Poloxamer 188 (mg) |
Foamability Volume (mL) |
Foam Stability (min) |
|
100 |
37.8 |
10.0 |
|
200 |
45.6 |
11.0 |
|
300 |
53.2 |
12.5 |
|
400 |
52.5 |
11.5 |
|
500 |
51.0 |
12.0 |
3.3 Evaluation of Floating Beads
3.3.1 Percentage Yield and Drug Entrapment Efficiency
The percentage yield and drug entrapment efficiency of all formulations (F1-F5) are presented in Table 6 and Figure 1.
Table 6: Percentage Yield and Drug Entrapment of Formulations F1-F5
|
S. No. |
Formulation code |
%Yield |
%Drug entrapment |
|
1 |
F1 |
38.09±0.003 |
25.22±0.005 |
|
2 |
F2 |
45.02±0.006 |
35.42±0.002 |
|
3 |
F3 |
72.52±0.005 |
73.69±0.003 |
|
4 |
F4 |
70.49±0.004 |
71.69±0.001 |
|
5 |
F5 |
56.14±0.005 |
69.56±0.005 |
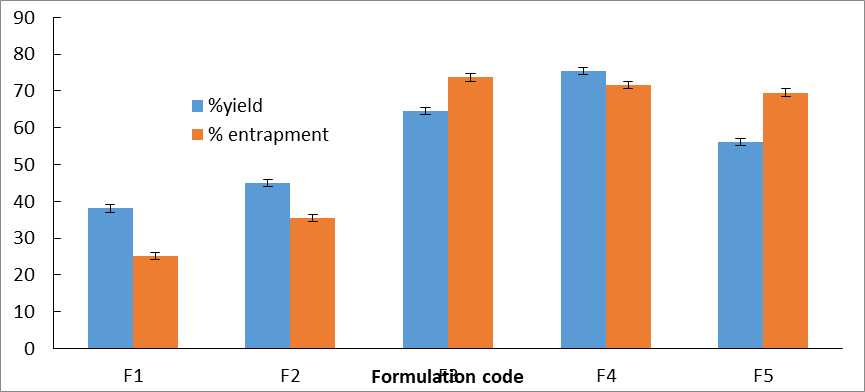
Fig 1: %Yield and drug entrapment of different formulation
Effect on Percentage Yield:
The percentage yield increased progressively with sodium alginate concentration up to 1.5% w/v (F3: 72.52%). This increase is attributed to the higher viscosity of the polymer solution, which facilitates formation of larger, more robust spheres and reduces loss during extrusion and curing [7]. However, further increase in alginate concentration to 2.5% (F5) resulted in decreased yield (56.14%) due to excessive viscosity, which hindered smooth droplet passage through the needle, leading to irregular particle formation and fragmentation during hardening.
Effect on Drug Entrapment Efficiency:
The drug entrapment efficiency increased significantly from 25.22% (F1) to 73.69% (F3) as alginate concentration increased from 0.5% to 1.5% w/v. At lower concentrations, the dilute polymer matrix is porous and insufficiently cross-linked, allowing drug diffusion into the surrounding calcium chloride solution. At optimal concentrations (F3 and F4), the denser alginate matrix effectively entraps the drug. The slight decrease at higher concentrations (F5: 69.56%) may be attributed to rapid surface gelation creating a barrier that slows inward calcium diffusion, resulting in a less uniformly cross-linked core.
3.3.2 In-vitro Floating Study
The floating behavior of all formulations is presented in Table 7. All formulations showed excellent floating properties with 100% buoyancy maintained for up to 7 hours, except F5 which showed 80-90% buoyancy at intermediate time points. The rapid floating of the beads is attributed to the porous structure created by the foaming agent, which lowers the density below that of gastric fluid (<1.004 g/cm³).
Table 7: Percentage Floating of Different Formulations
3.3.3 Selection of Optimized Formulation
Based on the evaluation of percentage yield and drug entrapment, Formulation F3 (1.5% w/v sodium alginate) was selected as the optimized formulation. It demonstrated the highest percentage yield (72.52%) and maximum drug entrapment (73.69%), indicating excellent process efficiency and drug loading capacity. Although F4 showed comparable results, F3 was preferred due to better yield and robust bead formation with lower polymer concentration.
3.4 In-vitro Drug Release Study
The in-vitro drug release profile of the optimized formulation F3 is presented in Table 10 and Figure 2.
Table 8: Percentage Drug Release from F3 Formulation
|
S. No. |
Time (hours) |
% Drug Release of F3 Formulation |
|
1 |
0 |
0±0 |
|
2 |
1 |
17.31±0.29 |
|
3 |
2 |
28.8±0.34 |
|
4 |
3 |
42.51±0.51 |
|
5 |
4 |
48.88±0.77 |
|
6 |
5 |
57.75±0.94 |
|
7 |
6 |
65.76±0.86 |
|
8 |
7 |
73.9±0.54 |
|
9 |
8 |
79.8±0.21 |
|
10 |
9 |
85.72±0.27 |
|
11 |
10 |
90.11±0.19 |
|
12 |
11 |
92.62±0.15 |
|
13 |
24 |
93.71±0.21 |
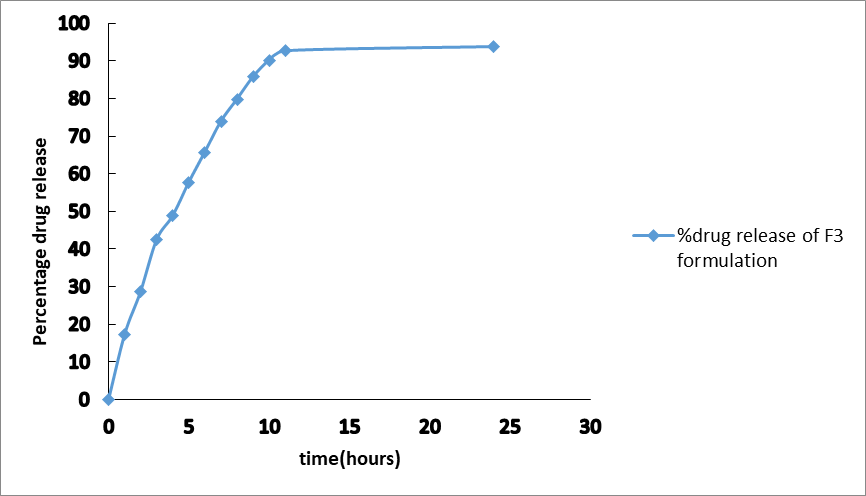
Fig 2: Percentage drug release of F3 formulation
The optimized formulation F3 showed a biphasic release pattern with an initial burst release of 17.31% in the first hour, followed by sustained release over 24 hours (93.71%). The initial burst release may be attributed to the rapid dissolution of drug molecules present on the surface of the porous beads. The sustained release thereafter is due to diffusion of the drug through the swollen alginate matrix [8].
3.5 Drug Release Kinetics
The in-vitro release data were fitted to various kinetic models to elucidate the release mechanism (Table 9).
Table 9: Kinetic Parameters of Formulation F3
|
Formulation name |
Zero order |
First order |
Higuchi |
Peppas |
||||
|
R2 |
K0 |
R2 |
K0 |
R2 |
K0 |
R2 |
K0 |
|
|
F3
|
0.628 |
3.920 |
0.786 |
-0.058 |
0.942 |
72.72 |
1.199 |
0.829 |
3.5 Drug Release Kinetics
The release data showed the highest linearity for the Higuchi model (R² = 0.942), indicating that drug release from the floating beads follows a diffusion-controlled mechanism where the cumulative drug release is proportional to the square root of time [9]. The zero-order model showed poor fit (R² = 0.628), confirming that drug release is not concentration-independent.
The Korsmeyer-Peppas model yielded an n value of 0.829, which is between 0.5 and 1.0, indicating non-Fickian (anomalous) diffusion mechanism. This suggests that drug release is governed by a combination of diffusion through the swollen polymer matrix and polymer chain relaxation (swelling-controlled release) [10].
DISCUSSION
The formulation of gastroretentive floating beads of Ramipril using the foaming technique demonstrated promising results. The pre-formulation studies confirmed the identity, purity, and physicochemical properties of the drug. The pH-dependent solubility of Ramipril justifies the need for a gastroretentive formulation, as the drug shows minimal solubility at gastric pH and better solubility at intestinal pH.
The optimization of Poloxamer 188 concentration revealed that 300 mg provided optimal foamability and foam stability. Poloxamer 188, being an amphiphilic surfactant, reduces surface tension and stabilizes the foam through orientation at the air-water interface [11]. The addition of sodium alginate further stabilized the foam through surfactant-polymer complex formation.
The ionotropic gelation technique using calcium chloride as the cross-linking agent successfully produced porous floating beads. The concentration of sodium alginate significantly influenced the formulation characteristics. The increase in drug entrapment with increasing alginate concentration up to 1.5% w/v can be attributed to the formation of a denser polymer matrix that effectively entraps the drug [12]. However, at higher concentrations, the excessive viscosity led to irregular bead formation and reduced yield.
The excellent floating properties observed for all formulations (100% buoyancy for up to 7 hours) confirm the effectiveness of the foaming technique in creating porous, low-density structures. The sustained release of Ramipril from the optimized formulation over 24 hours indicates the potential of these beads to maintain therapeutic drug levels for prolonged periods.
The release kinetics following the Higuchi model and the n value indicating non-Fickian diffusion suggest that drug release is controlled by both diffusion and polymer relaxation mechanisms. The porosity created by the foaming agent facilitates initial water penetration and drug dissolution, while the alginate matrix provides a barrier for controlled release.
Compared to conventional immediate-release formulations of Ramipril, these floating beads offer several advantages: prolonged gastric residence time, sustained drug release, reduced dosing frequency, and improved patient compliance [13]. The multi-particulate nature of the beads also reduces the risk of dose dumping and provides more predictable pharmacokinetics [14].
CONCLUSION
The present study successfully developed and evaluated gastroretentive floating beads of Ramipril using the foaming technique. The pre-formulation studies confirmed the purity and physicochemical properties of the drug. Poloxamer 188 at 300 mg was optimized as the foaming agent based on foamability and foam stability studies.
Among the five formulations prepared with varying sodium alginate concentrations, Formulation F3 (1.5% w/v sodium alginate) was identified as the optimized formulation based on higher percentage yield (72.52%) and maximum drug entrapment (73.69%). All formulations showed excellent floating properties, maintaining 100% buoyancy for up to 7 hours.
The in-vitro drug release from F3 showed sustained release of 93.71% over 24 hours, following Higuchi diffusion-controlled kinetics with non-Fickian release mechanism (n=0.829). The porous structure created by the foaming agent facilitated initial drug release, while the alginate matrix provided sustained release characteristics.
The developed floating beads demonstrate promising potential for improving the bioavailability and therapeutic efficacy of Ramipril through prolonged gastric retention and controlled drug release. These formulations could significantly reduce dosing frequency, minimize peak-and-valley plasma concentration fluctuations, and improve patient compliance in the management of hypertension.
REFERENCES


Hitesh Yadav, Nikita Kaushik, Dr Parveen Kumari, Formulation And Evaluation of Gastroretentive Floating Beads of Ramipril Using Foaming Technique, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 7, 636-645, https://doi.org/10.5281/zenodo.21156494
 10.5281/zenodo.21156494
10.5281/zenodo.21156494