
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutical Quality Assurance, S.S.P. Shikshan Sanstha’s Siddhi College of Pharmacy, Chikhali, Pune, Maharashtra, India - 411062
One of the most prevalent dermatological conditions affecting the skin and mucosal tissues globally is superficial fungal infections. Traditional topical antifungal formulations frequently show diminished therapeutic efficiency, short retention times, and inadequate skin penetration. The goal of the current study was to create and assess a liposomal gel loaded with tioconazole for improved topical antifungal activity. Although tioconazole is a broad-spectrum imidazole antifungal drug, its limited penetration into the stratum corneum and poor aqueous solubility limit its efficacy in topical therapy. In order to get around these restrictions, soy lecithin and cholesterol were used as lipid components in the thin-film hydration process to create tioconazole-loaded liposomes, which were then added to the Carbopol 940 gel basis. Physical characteristics, pH, viscosity, spreadability, drug content, entrapment effectiveness, vesicle size, zeta potential, in vitro drug diffusion, ex vivo permeation, antifungal efficacy, and stability tests were all assessed for the produced formulations. With acceptable homogeneity, an appropriate pH, high entrapment efficiency, and nanosized vesicles, the improved formulation demonstrated satisfactory physicochemical attributes. Studies on ex-vivo penetration and in-vitro diffusion showed improved skin penetration and prolonged drug release when compared to traditional gel formulation. Because of its enhanced penetration and extended drug retention at the infection site, the liposomal gel also demonstrated higher antifungal effectiveness against Candida albicans. The formulation remained stable under the recommended storage conditions, according to stability studies. According to the current study's findings, tioconazole liposomal gel may be a viable topical medication delivery method for treating superficial fungal infections with better therapeutic efficacy, longer drug release, increased patient compliance, and fewer doses.
Millions of people worldwide suffer from superficial fungal infections, which are among the most common dermatological conditions. Dermatophytes, yeasts, and molds that infiltrate keratinized tissues like skin, hair, and nails are the primary culprits behind these illnesses. A number of factors, including humid environments, immunocompromised states, diabetes, prolonged antibiotic medication, and poor hygiene habits, have contributed to a large increase in the frequency of fungal infections. Topical antifungal therapy is still the recommended treatment for superficial mycoses, but traditional formulations, such as creams, ointments, and lotions, frequently have drawbacks like poor skin penetration, short residence times, poor retention at the target site, and frequent application requirements, which may ultimately lower patient compliance and therapeutic efficacy [1, 2]
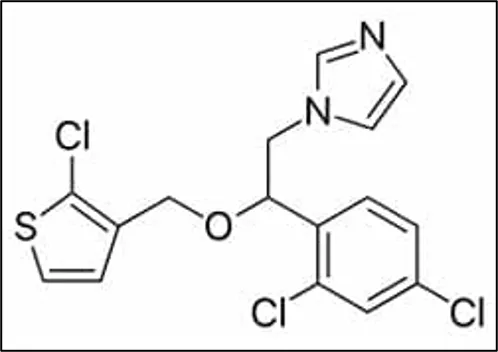
Figure 1 Structure of Tioconazole
Tioconazole is a synthetic imidazole derivative with broad-spectrum antifungal activity against dermatophytes, yeasts, and other pathogenic fungi. Its IUPAC name is -[2-[(2-Chlorothiophen-3-yl)methoxy]-2-(2,4-dichlorophenyl)ethyl]-1H-imidazole ( Figure 1), its molecular formula is C₁₆H₁₃Cl₃N₂OS, and its molecular weight is 387.71g/mol [3]. It increases membrane permeability and causes fungal cell death by preventing the formation of ergosterol, a crucial part of the fungal cell membrane [4]. Although tioconazole is frequently used to treat superficial fungal infections, its low penetration through the stratum corneum and poor water solubility limit its therapeutic efficacy. These difficulties call for the creation of a cutting-edge topical delivery system that can improve medication penetration and retention in the skin's layers [5].
Innovative vesicular drug delivery methods have demonstrated significant promise in addressing the drawbacks of traditional topical formulations. Because of their biocompatibility, biodegradability, and capacity to encapsulate both hydrophilic and lipophilic medications, liposomes have drawn a lot of interest. By increasing medication localization within the skin and regulating drug release, liposomes—microscopic phospholipid vesicles—can improve dermal and transdermal drug delivery. Additionally, their structural similarity to biological membranes makes it easier for them to interact with the skin, which improves penetration and lessens local irritation [6, 7].
The incorporation of liposomal vesicles into a gel matrix offers additional advantages for topical application. Liposomal gels provide improved viscosity, spreadability, patient acceptability, and prolonged contact time at the site of application. Such systems may also enhance drug stability and improve therapeutic efficacy by maintaining sustained drug release and increasing localized drug concentration [8]. Therefore, the combination of liposomes with gel-based systems represents a promising strategy to anti fungle activity.
Few research have particularly examined tioconazole-loaded liposomal gel formulations for increased topical antifungal activity, despite the fact that numerous vesicular formulations have been studied for antifungal drug delivery. In order to enhance drug entrapment efficiency, skin penetration, drug release properties, and antifungal activity, the current work attempts to develop and assess a tioconazole liposomal gel. For the treatment of superficial fungal infections, the proposed formulation might be a useful substitute for traditional topical antifungal medications.
According to recent research, topical antifungal therapy is greatly improved by vesicular drug delivery methods like liposomes, which increase skin penetration, drug retention, and prolonged release. Antifungal medications can be encapsulated in liposomes, which are biocompatible phospholipid vesicles that enhance their therapeutic efficacy. The benefits of liposomal-based systems in enhancing drug penetration and lowering adverse effects related to topical antifungal therapy were demonstrated by published literature reviews [9-15]. Additionally, researchers verified that one of the most popular and successful techniques for liposome preparation is thin-film hydration. Tioconazole-loaded liposomal gel formulations have not, however, been the subject of any research, which highlights the necessity of the current investigation.
A free sample of tioconazole was acquired from Themis Medicare Pvt. Ltd. in Haridwar, India. To make liposomes, soy lecithin and cholesterol were utilized as lipid components. Propylene glycol served as a humectant and penetration enhancer, and carbopol 940 was utilized as the gelling agent. To increase the formulation's moisturizing and spreadability, glycerin was added. Triethanolamine was utilized for pH correction and gel base neutralization, whereas methyl and propyl paraben were utilized as preservatives. Methanol and chloroform were employed as organic solvents to produce lipid films. During in vitro experiments, phosphate buffer pH 7.4 was used as a hydration and diffusion medium. Every chemical and reagent utilized in the research was of analytical quality.
Tioconazole's physicochemical characteristics and compatibility with the formulation's excipients were assessed using preformulation tests. The creation of a stable and efficient dosage form depends on these investigations.
The medication sample's color, odor, texture, and overall look were all visually assessed. To verify the drug's identification and purity, the observations were verified with regulatory standards.
Distilled water, methanol, ethanol, phosphate buffer pH 7.4, and chloroform were among the solvents in which the solubility of tioconazole was assessed. For a full day at room temperature, an excess amount of medication was put to each solvent and continually shaken. To ascertain the drug solubility, the solutions were filtered and subjected to spectrophotometric analysis.
The capillary fusion method was used to measure the melting point of tioconazole.[16] A capillary tube with one end capped was filled with a tiny amount of medication and put into a melting point equipment. The drug's melting temperature range was noted and compared to published values.
Fourier Transform Infrared (FTIR) spectroscopy was used to conduct drug-excipient compatibility investigations. A range of 4000–400 cm−1 was used to record the FTIR spectra of the improved formulation, physical mixes, and pure medication. The spectra were examined for distinctive peaks and potential drug-excipient interactions.
The composition of liposomes formulation as per given Table 1
Table 1 Composition of Tioconazole Liposomes
|
Ingredients |
F1 |
F2 |
F3 |
F4 |
|
Tioconazole (g) |
1 |
1 |
1 |
1 |
|
Soya Lecithin (g) |
2 |
3 |
4 |
5 |
|
Cholesterol (g) |
0.5 |
1 |
1.5 |
2 |
|
Chloroform: Methanol |
q.s. |
q.s. |
q.s. |
q.s. |
|
Phosphate Buffer pH 7.4 |
q.s. |
q.s. |
q.s. |
q.s. |
Tioconazole-loaded liposomes were prepared by thin-film hydration technique [17] using rotary vacuum evaporation method. [18, 19]
Step 1: Lipid Solution Preparation
In a dry, clean round-bottom flask, precisely weighed amounts of tioconazole, soy lecithin, and cholesterol were dissolved in a 2:1 v/v chloroform: methanol combination. To create a clear, uniform solution, the lipid solution was thoroughly mixed.
Step 2: Thin Lipid Film Formation
A rotating vacuum evaporator was connected to the round-bottom flask holding the lipid solution. At 40–45°C and 100–120 rpm, the organic solvent was evaporated under low pressure until a thin, homogeneous lipid coating developed on the flask's inner wall. To guarantee that all leftover solvent was removed, the flask was placed in a desiccator for the whole night
Step 3: Hydration of Lipid Film
Phosphate buffer pH 7.4 was used to hydrate the dried lipid film at a temperature higher than the lipid transition temperature. To create multilamellar vesicles, hydration was maintained for about an hour while rotating continuously.
Step 4: Sonication
To create tiny unilamellar vesicles with a limited size distribution, the generated liposomal dispersion was sonicated for five to ten minutes using a probe sonicator.
Step 5: Unentrapped Drug Separation
For thirty minutes, the liposomal suspension was centrifuged at 12,000 rpm. After separating the free drug-containing supernatant, the sedimented liposomes were gathered and redistributed in phosphate buffer.
The gel base was prepared using Carbopol 940 and subsequently incorporated with optimized liposomal dispersion. Table 2
Table 2 Composition of Liposomal Gel
|
Ingredients |
Quantity (%) |
|
Tioconazole-loaded liposomes |
Equivalent to 1% drug |
|
Carbopol 940 |
1 |
|
Propylene glycol |
7 |
|
Glycerin |
5 |
|
Methyl paraben |
0.1 |
|
Propyl paraben |
0.02 |
|
Triethanolamine |
q.s. |
|
Purified water |
q.s. to 100 g |
Step 1: Gel Base Preparation
To avoid lump formation, carbopol 940 was gradually distributed in purified water while being continuously stirred mechanically. To fully hydrate and expand the polymer, the dispersion was left to stand for three to four hours.
Step 2: Including Additives
Glycerin and propylene glycol were added to the hydrated gel basis while being constantly stirred. As preservatives, methyl and propyl parabens were individually dissolved and added to the gel basis.
Step 3: Neutralization
Triethanolamine was gradually added to the mixture while being gently stirred until a translucent gel with the right pH and viscosity was achieved.
Step 4: Liposome Incorporation
To create a homogenous liposomal gel, the produced tioconazole liposomal dispersion was gradually added to the gel basis while being gently stirred. To prevent liposomal vesicle rupture, care was made to minimize severe agitation.
With constant stirring, carbopol 940 was gradually dissolved in purified water and given time to fully hydrate. The hydrated gel base was supplemented with propylene glycol, glycerin, methyl paraben, and propyl paraben. Drop by drop, triethanolamine was added until a clear gel was achieved. To create a homogenous tioconazole liposomal gel, the prepared liposomal dispersion was then gently stirred into the gel basis.
Numerous physicochemical and performance metrics were assessed for the generated liposomal formulations and liposomal gel.
The color, homogeneity, consistency, phase separation, and grittiness of the produced formulations were visually examined.
After dispersing one gram of gel in one hundred milliliters of distilled water, the mixture was let to stand for two hours. A digital pH meter that had been calibrated was used to measure the pH at room temperature. Three separate measurements were made.
The resulting gel compositions' viscosity was tested using a Brookfield viscometer with an appropriate spindle rotating at a set speed.
Glass slide equipment was used to measure spreadability. A predetermined weight was used to compress the extra gel between two glass slides. The time required for the upper slide to move a specified distance was recorded.
A precisely measured amount of gel equal to 10 mg of medication was dissolved in methanol and suitably diluted. A UV-visible spectrophotometer was used to filter the fluid and perform spectrophotometric analysis.
Entrapment efficiency was determined by centrifugation method. Liposomal dispersion was centrifuged and free drug present in supernatant was estimated spectrophotometrically.
Formula for Entrapment Efficiency
Entrapment Efficiency (%)=Total Drug-Free DrugTotal Drug×100
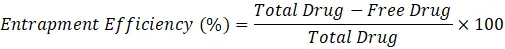
Particle size, polydispersity index, and zeta potential of liposomes were determined using dynamic light scattering method.
In-vitro diffusion study was performed using Franz diffusion cell fitted with dialysis membrane. Phosphate buffer pH 7.4 was used as receptor medium maintained at 37 ± 0.5°C with continuous stirring. Samples were withdrawn at predetermined intervals and analyzed spectrophotometrically.
Antifungal activity was evaluated against Candida albicans using cup-plate method. Zones of inhibition produced by the formulations were measured and compared with conventional formulation.
Stability studies were performed according to ICH guidelines [20, 21] at refrigerated temperature and room temperature for a specified period. Formulations were evaluated periodically for physical appearance, pH, drug content, and entrapment efficiency.
The prepared tioconazole liposomal formulations were successfully developed and evaluated for various physicochemical and performance parameters. The obtained results indicated that liposomal incorporation significantly improved topical delivery characteristics of tioconazole.
3.1. Preformulation Studies
3.1.1. Organoleptic properties
The organoleptic properties of Tioconazole were evaluated for color, odor, appearance, and physical nature. The drug was found to comply with standard organoleptic characteristics. Table 3
Table 3 Organoleptic properties of drug
|
Parameter |
Observation |
|
Color |
White to off-white |
|
Odor |
Characteristic |
|
Appearance |
Crystalline powder |
|
Physical Nature |
Fine and free-flowing powder |
3.1.2 Solubility
The solubility of Tioconazole was determined in different solvents at room temperature. The drug exhibited poor solubility in water and good solubility in organic solvents. Table 4
Table 4 Solubility of Drug
|
Solvent |
Solubility Observation |
|
Distilled Water |
Practically insoluble |
|
Phosphate Buffer pH 7.4 |
Slightly soluble |
|
Ethanol |
Soluble |
|
Methanol |
Freely soluble |
|
Chloroform |
Freely soluble |
3.1.3. Melting Point
The melting point of Tioconazole was determined by capillary fusion method using melting point apparatus. The observed melting point range was found to be in accordance with reported standard values. Table 5
Table 5 Melting point of drug
|
Parameter |
Observation |
|
Melting Point |
82°C – 86°C |
3.1.4. Drug-Excipient compatibility test
The compatibility study between Tioconazole and selected excipients was carried out using Fourier Transform Infrared (FTIR) spectroscopy. FTIR spectra of pure drug, physical mixture, and optimized liposomal gel formulation were analyzed for characteristic functional peaks. Table 6, Figure 2
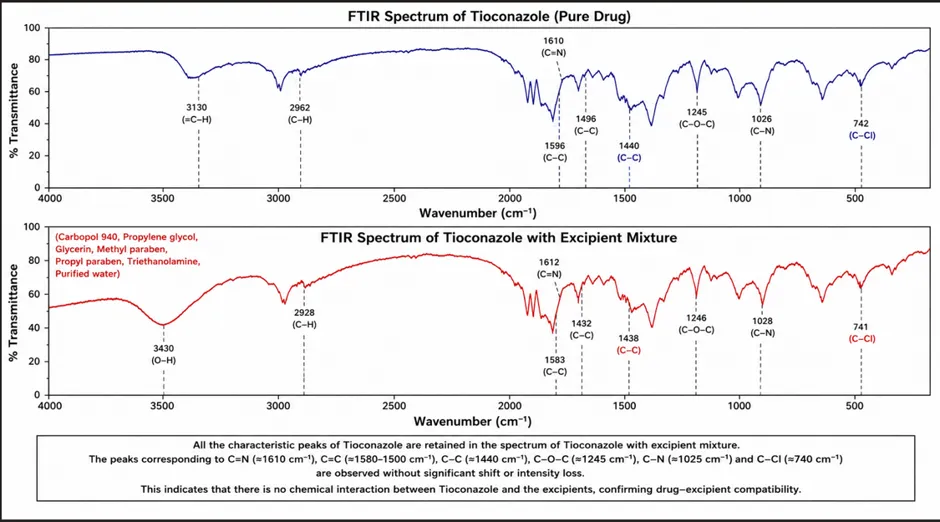
Figure 2 FTIR spectra of Drug-Excipient compatibility
Table 6 Observed FTIR Peaks of Tioconazole
|
Functional Group |
Characteristic Peak (cm⁻¹) |
Observation |
|
C–Cl stretching |
740–760 |
Present |
|
C=N stretching |
1580–1620 |
Present |
|
Aromatic C=C stretching |
1450–1500 |
Present |
|
C–O stretching |
1100–1250 |
Present |
|
C–H stretching |
2850–3100 |
Present |
The characteristic peaks of tioconazole were retained in the optimized formulation without significant shifting, disappearance, or formation of new peaks.
3.2. Tioconazole Liposomal gel formulation evaluation parameters
3.2.1. Physical Appearance
The prepared Tioconazole liposomal gel formulations were visually examined for color, homogeneity, consistency, grittiness, and phase separation. All formulations were found to be smooth, homogeneous, and free from visible particulate matter or phase separation. Table 7
Table 7 Physical appearance of gel
|
Formulation |
Color |
Homogeneity |
Consistency |
Grittiness |
Phase Separation |
|
F1 |
White |
Good |
Smooth |
Absent |
Absent |
|
F2 |
Off-white |
Good |
Smooth |
Absent |
Absent |
|
F3 |
White to off-white |
Excellent |
Smooth and uniform |
Absent |
Absent |
|
F4 |
Slightly creamy white |
Good |
Slightly viscous |
Absent |
Absent |
The optimized formulation (F3) showed excellent homogeneity with smooth texture and elegant appearance suitable for topical application.
3.2.2. pH determination
The pH of the prepared Tioconazole liposomal gel formulations was found to be within the range of 6.1 ± 0.2 to 6.8 ± 0.1, indicating suitability for topical application. The optimized formulation (F3) showed a pH of 6.4 ± 0.2, which is close to the normal pH of human skin. Table 8
Table 8 pH of gel
|
Formulation |
pH (Mean ± SD, n = 3) |
|
F1 |
6.1 ± 0.2 |
|
F2 |
6.3 ± 0.1 |
|
F3 |
6.4 ± 0.2 |
|
F4 |
6.8 ± 0.1 |
3.2.3. Viscosity
The viscosity of the prepared Tioconazole liposomal gel formulations was measured using a Brookfield viscometer at room temperature. All formulations exhibited satisfactory viscosity suitable for topical application. Table 9
Table 9 Viscosity of gel
|
Formulation |
Viscosity (cps) (Mean ± SD, n = 3) |
|
F1 |
3560 ± 18 |
|
F2 |
3895 ± 22 |
|
F3 |
4120 ± 24 |
|
F4 |
4386 ± 20 |
The optimized formulation (F3) showed a viscosity of 4120 ± 24 cps, which provided suitable consistency, ease of application, and good spreadability.
3.2.4. Spreadability
The spreadability of the prepared Tioconazole liposomal gel formulations was determined using the glass slide method. All formulations exhibited satisfactory spreadability indicating ease of application on the skin surface. Table 10
Table 10 spreadability of gel
|
Formulation |
Spreadability (g·cm/sec) (Mean ± SD, n = 3) |
|
F1 |
12.4 ± 0.3 |
|
F2 |
14.1 ± 0.4 |
|
F3 |
16.3 ± 0.2 |
|
F4 |
13.8 ± 0.5 |
The optimized formulation (F3) showed maximum spreadability of 16.3 ± 0.2 g·cm/sec, indicating smooth application and uniform spreading characteristics.
3.2.5. Drug content
The drug content of the prepared Tioconazole liposomal gel formulations was determined using UV-visible spectrophotometric analysis. All formulations showed satisfactory drug content indicating uniform distribution of drug within the gel matrix. Table 11
Table 11 Drug content of gel
|
Formulation |
Drug Content (%) (Mean ± SD, n = 3) |
|
F1 |
94.26 ± 0.6 |
|
F2 |
96.14 ± 0.5 |
|
F3 |
98.12 ± 0.8 |
|
F4 |
97.03 ± 0.7 |
The optimized formulation (F3) exhibited maximum drug content of 98.12 ± 0.8%, indicating efficient incorporation and uniform dispersion of tioconazole within the liposomal gel system.
3.2.6. Entrapment efficiency
Centrifugation was used to measure the entrapment efficiency of Tioconazole liposomes, and the unentrapped medication was then subjected to UV spectrophotometric measurement. Tioconazole was successfully incorporated into liposomal vesicles in all formulations, as evidenced by acceptable drug entrapment. Table 12
Table 12 Entrapment efficiency of gel
|
Formulation |
Entrapment Efficiency (%) (Mean ± SD, n = 3) |
|
F1 |
72.18 ± 0.5 |
|
F2 |
78.64 ± 0.7 |
|
F3 |
85.42 ± 0.6 |
|
F4 |
81.27 ± 0.8 |
The optimized formulation (F3) showed maximum entrapment efficiency of 85.42 ± 0.6%, indicating efficient encapsulation of tioconazole into the lipid vesicles. Figure 3
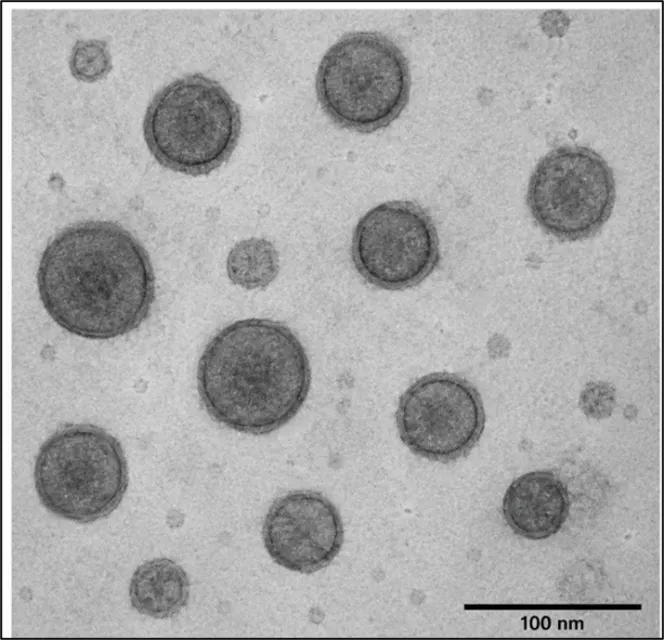
Figure 3 Transmission Electron Microscopy (TEM) of Tioconazole Liposomes of F3
3.2.7. Vesicle size and Zeta potential
The vesicle size and zeta potential of the optimized Tioconazole liposomal formulation (F3) were determined using dynamic light scattering (DLS) technique. The formulation exhibited nanosized vesicles with good surface charge indicating stability of the liposomal dispersion. Table 13
Table 13 vesicle size and zeta potential of optimized gel formulation
|
Parameter |
Observation |
|
Vesicle Size |
186.4 ± 4.2 nm |
|
Polydispersity Index (PDI) |
0.236 ± 0.02 |
|
Zeta Potential |
–32.5 ± 1.4 mV |
The optimized formulation showed uniform nanosized vesicles with narrow size distribution and adequate zeta potential for physical stability.
3.2.8. In-vitro Drug Diffusion Study
The in-vitro drug diffusion study of Tioconazole liposomal gel formulations was carried out using Franz diffusion cell containing phosphate buffer pH 7.4 as diffusion medium. The cumulative percentage drug release was measured over a period of 12 hours. Table 14
Table 14 Cumulative Drug Release Data
|
Time (h) |
F1 (%) |
F2 (%) |
F3 (%) |
F4 (%) |
|
1 |
18.2 ± 0.4 |
20.4 ± 0.5 |
24.6 ± 0.3 |
21.3 ± 0.4 |
|
2 |
28.5 ± 0.5 |
34.2 ± 0.4 |
39.8 ± 0.6 |
35.1 ± 0.5 |
|
4 |
42.3 ± 0.6 |
50.6 ± 0.5 |
58.7 ± 0.4 |
53.2 ± 0.6 |
|
6 |
55.8 ± 0.4 |
64.1 ± 0.7 |
72.4 ± 0.5 |
67.5 ± 0.5 |
|
8 |
66.9 ± 0.5 |
75.8 ± 0.6 |
84.3 ± 0.4 |
79.1 ± 0.7 |
|
10 |
74.6 ± 0.7 |
83.5 ± 0.5 |
91.2 ± 0.6 |
86.4 ± 0.5 |
|
12 |
81.3 ± 0.5 |
89.7 ± 0.4 |
96.8 ± 0.5 |
91.5 ± 0.6 |
The optimized formulation (F3) showed maximum cumulative drug release of 96.8 ± 0.5% after 12 hours.
3.2.9. Antifungal Activity
The antifungal activity of the prepared Tioconazole liposomal gel formulations was evaluated by cup-plate (agar well diffusion) method using Candida albicans as test organisms. The zone of inhibition produced by the formulations was compared with marketed Miconazole gel and drug suspension. Table 15 and Figure 4
Table 15 Zone of Inhibition
|
Formulation |
Candida albicans (mm) |
|
Blank Gel |
0.0 ± 0.0 |
|
Marketed Gel (Miconazole 1% w/w) |
14.2 ± 0.6 |
|
Tioconazole Suspension (1% w/w) |
18.6 ± 0.5 |
|
Tioconazole Liposomal Gel (F2) |
21.3 ± 0.6 |
|
Tioconazole Liposomal Gel (F3) |
26.7 ± 0.5 |
The optimized formulation (F3) showed the highest zone of inhibition against both fungal strains, indicating superior antifungal activity.
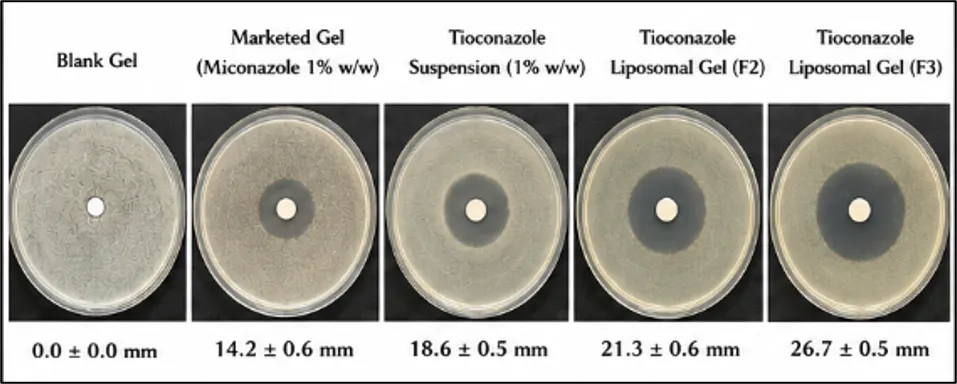
Figure 4 Zone of Inhibition against Candida albicans
3.6 Stability Studies
3.2.10. Stability study
In accordance with International Council for Harmonization criteria, a three-month stability study of the optimized Tioconazole liposomal gel formulation (F3) was conducted at room temperature (25 ± 2°C/60 ± 5% RH) and in a refrigerator (4 ± 2°C). The formulation's physical characteristics, pH, drug content, and entrapment effectiveness were all routinely assessed. Table 16
Table 16 Stability Study Data of Optimized Formulation (F3)
|
Parameter |
Initial |
After 1 Month |
After 2 Months |
After 3 Months |
|
Physical Appearance |
Smooth and homogeneous |
No change |
No change |
No change |
|
pH |
6.4 ± 0.2 |
6.3 ± 0.1 |
6.3 ± 0.2 |
6.2 ± 0.1 |
|
Drug Content (%) |
98.12 ± 0.8 |
97.54 ± 0.6 |
96.88 ± 0.5 |
96.14 ± 0.7 |
|
Entrapment Efficiency (%) |
85.42 ± 0.6 |
84.96 ± 0.5 |
84.18 ± 0.7 |
83.52 ± 0.6 |
No significant changes were observed in physical appearance, pH, drug content, or entrapment efficiency during the study period.
The current work effectively created and assessed a liposomal gel filled with tioconazole for topical antifungal treatment. The proposed formulation showed better skin penetration, longer drug release, improved entrapment efficiency, and superior antifungal activity in addition to good physicochemical properties. Liposomes were successfully included into the gel basis to improve tioconazole topical administration. For the successful treatment of superficial fungal infections, tioconazole liposomal gel may therefore be a viable substitute for traditional topical antifungal formulations.
ACKNOWLEDGEMENT
We acknowledge our sincere thanks to S.S.P. Shikshan Sanstha’s Siddhi College of Pharmacy, Chikhali, Pune for providing facilities to carryout research work.
DECLARATIONS
Author Contribution: Authors Arti Ingole, Hitanshi Darji, Ashwini Pansare performed the research and wrote the main manuscript and Author Pravin Sable reviewed the manuscript.
Funding: Not applicable
Data Availability: Data will be made available on request.
Ethics approval: Not applicable
Consent to participate: Not applicable
Consent for publication: Not applicable
Competing interests: The authors declare no competing interests.
REFERENCES




Arti Ingole, Hitanshi Darji, Ashwini Pansare, P. N. Sable, Formulation and Evaluation of Tioconazole Liposomal Gel for Enhanced Topical Antifungal Activity, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 5759-5770. https://doi.org/10.5281/zenodo.20801417
 10.5281/zenodo.20801417
10.5281/zenodo.20801417