
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutical Analysis, National College of Pharmacy.
Pharmaceuticals represent a rapidly expanding class of contaminants of emerging concern (CECs) detected worldwide in surface water, groundwater, wastewater, and drinking water, raising profound ecological and public health concerns. Conventional analytical methods used for their detection rely on large volumes of hazardous organic solvents, energy-intensive instrumentation, and complex sample preparation procedures that conflict with the principles of green analytical chemistry (GAC). This review critically examines environmentally responsible approaches for the detection of pharmaceutical contaminants in diverse aquatic matrices. A comprehensive evaluation of green sampling strategies—including passive sampling with POCIS and Chemcatcher devices—is presented alongside miniaturised sample preparation techniques such as solid-phase microextraction (SPME), dispersive liquid-liquid microextraction (DLLME), and greener solid-phase extraction (SPE) formats incorporating novel sorbents including molecularly imprinted polymers (MIPs) and metal-organic frameworks (MOFs). Advances in ultra-high-performance liquid chromatography (UHPLC) coupled to tandem mass spectrometry (MS/MS) and high-resolution mass spectrometry (HRMS) are discussed in the context of improving sensitivity while reducing analytical footprint. Greenness assessment tools—the Analytical Eco-Scale, GAPI, and AGREE—are evaluated and compared. A decision framework is proposed to guide method selection by matrix type, target compound class, and laboratory resources. Critical knowledge gaps are identified, including the need for standardised greenness reporting, lifecycle assessment of novel sorbents, and validation of portable field-deployable analytical platforms. The review concludes that genuinely greener approaches are achievable without sacrificing detection performance when selection is guided by both analytical and sustainability criteria
Contaminants of emerging concern (CECs) encompass a broad and structurally diverse range of synthetic chemicals including pharmaceuticals and personal care products (PPCPs), pesticides, per- and polyfluoroalkyl substances (PFASs), micro- and nanoplastics, and engineered nanomaterials, all of which pose threats to environmental and human health (1, 2). These pollutants originate primarily from industrial activities, municipal wastewater effluents, household waste streams, and agricultural runoff, and are increasingly recognised as a new class of micropollutants that contaminate aquatic environments globally (3). Unlike conventional priority pollutants, CECs are often not subject to regulatory monitoring, and their environmental fate, transformation products, and mixture toxicity remain incompletely characterised (4).
Pharmaceutical compounds represent one of the most analytically and toxicologically significant subgroups within the CECs. They reach surface water and groundwater via multiple pathways: urban wastewater laden with pharmaceuticals from human excretion and improper disposal of unused or expired medicines; agricultural and livestock effluents from animals fed medicated feed whose excreta are applied to land as soil amendments; and discharges from pharmaceutical manufacturing facilities, where local contamination hotspots with exceptionally high drug concentrations have been documented in Asia, Europe, and North America despite stringent regulatory frameworks governing production (5, 6). Once released, pharmaceuticals are biologically active, pseudo-persistent, and frequently resistant to conventional wastewater treatment processes, enabling their continuous low-level introduction into receiving water bodies (7, 8).
Even at trace concentrations in the ng/L to low μg/L range, pharmaceuticals can exert chronic toxicity, endocrine disruption, and bioaccumulation in aquatic organisms (9, 10). The feminisation of fish populations due to oestrogenic compounds such as ethinylestradiol is among the most extensively documented examples of pharmaceutical ecotoxicity (11). Antimicrobial resistance (AMR) selection pressure from sub-therapeutic antibiotic concentrations in aquatic environments represents another critical concern with global public health implications (12). The pseudo-persistent nature of pharmaceutical contamination, driven by continuous input exceeding environmental degradation rates, means that exposure is effectively chronic even when individual compounds have relatively short half-lives (13).
Routine monitoring of pharmaceutical contaminants in aquatic systems is inherently challenging: they occur at trace and ultra-trace concentrations requiring highly sensitive and expensive analytical techniques; environmental samples are complex matrices containing organic matter, salts, and interfering co-contaminants; the structural diversity of hundreds of pharmaceutical compounds complicates simultaneous multi-residue analysis; and pharmaceuticals frequently exist as mixtures of parent compounds, metabolites, and transformation products, increasing analytical uncertainty (14, 15). Conventional methods for pharmaceutical detection are furthermore not considered green, relying on large organic solvent volumes, extensive sample preparation, and energy-intensive instrumentation including HPLC and LC-MS systems that generate substantial chemical waste and contradict the principles of GAC (16, 17).
Green Analytical Chemistry formally extends the 12 principles of Green Chemistry to analytical workflows, demanding minimisation of hazardous reagents, reduction of waste generation, energy efficiency, and operator safety without compromise of data quality (18). Its application to pharmaceutical water monitoring is not merely aspirational but operationally necessary given the scale of monitoring programmes required to address the global contamination challenge. This review therefore focuses on environmentally responsible detection of pharmaceuticals in aquatic matrices, covering sampling, sample preparation, separation, and detection, and integrates greenness assessment tools with analytical performance criteria to provide a practically actionable framework for researchers and monitoring laboratories (19, 20).
Pharmaceuticals in Aquatic Environments: Sources, Occurrence, and Analytical Targets
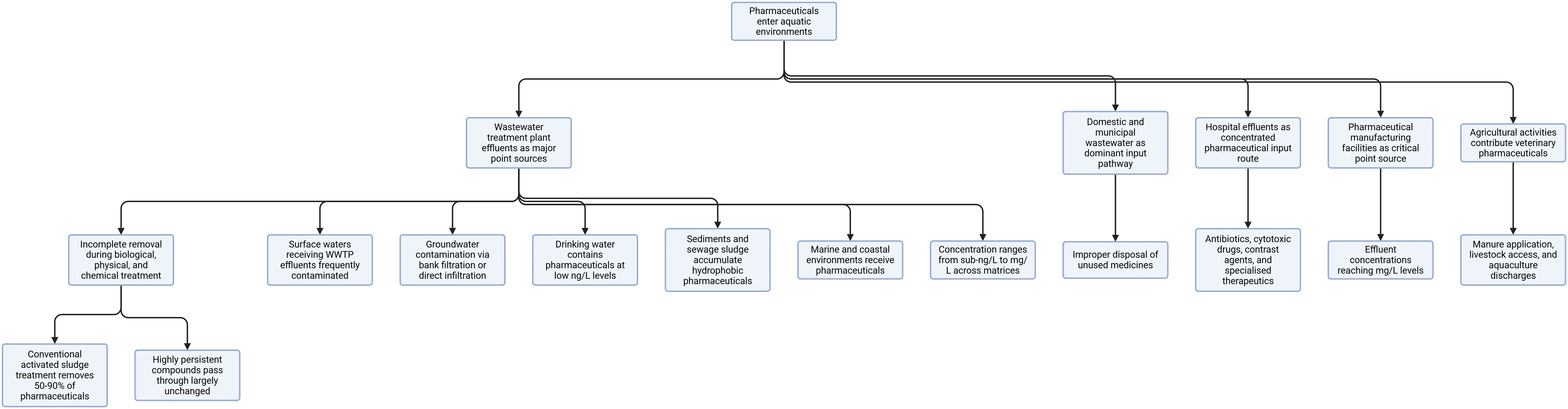
Pharmaceuticals enter aquatic environments primarily through wastewater treatment plant (WWTP) effluents, which act as major point sources due to incomplete removal of drug residues during biological, physical, and chemical treatment processes (21). Conventional activated sludge treatment removes many pharmaceuticals by 50-90%, but highly persistent compounds such as carbamazepine, diclofenac, and certain fluoroquinolone antibiotics pass through WWTPs largely unchanged (22). Domestic and municipal wastewater, enriched with pharmaceuticals excreted by humans in conjugated or unconjugated form and augmented by improper disposal of unused medicines via toilets and household waste, represents the dominant input pathway in urban settings (23).
Hospital effluents constitute a particularly concentrated pharmaceutical input route, introducing antibiotics, cytotoxic drugs, contrast agents, and specialised therapeutics at concentrations orders of magnitude higher than those in municipal wastewater (24). Studies from European and Asian hospital effluents have reported antibiotic concentrations in the μg/L range, with implications for AMR gene proliferation in receiving environments (25). Pharmaceutical manufacturing facilities represent another critical point source: surveys from India, China, and Europe have documented effluent concentrations reaching mg/L levels for specific drugs at production sites, overwhelmingly exceeding concentrations attributable to human excretion alone (26). Agricultural activities contribute veterinary pharmaceuticals to surface and groundwater through manure application, direct livestock access to water bodies, and aquaculture discharges, with tetracyclines, sulphonamides, and macrolide antibiotics being the most frequently detected classes from this pathway (27).
The occurrence of pharmaceuticals across different water body types reflects this diversity of sources. Surface waters receiving WWTP effluents are most frequently contaminated, with NSAIDs (diclofenac, ibuprofen, naproxen), antibiotics (ciprofloxacin, sulfamethoxazole, trimethoprim), antiepileptics (carbamazepine), antidepressants (fluoxetine, sertraline), beta-blockers (metoprolol, atenolol), lipid regulators (gemfibrozil, clofibric acid), and caffeine among the most commonly reported compounds globally (28, 29). Groundwater contamination via bank filtration or direct infiltration has been documented in multiple countries, with carbamazepine and sulfamethoxazole being particularly persistent indicators of wastewater influence due to their recalcitrance to soil attenuation (30). Drinking water contains pharmaceuticals at low ng/L levels in systems relying on surface water sources, with ibuprofen, carbamazepine, and atenolol among reported compounds; although concentrations are far below therapeutic thresholds, concerns about chronic low-level exposure, mixture effects, and inadequate toxicological understanding remain (31, 32).
Sediments and sewage sludge accumulate hydrophobic pharmaceuticals, particularly fluoroquinolone antibiotics and certain hormones, at concentrations substantially higher than those in overlying water due to strong sorption to organic matter and clay minerals (33). Their detection requires dedicated solid-matrix extraction approaches and represents a significant exposure reservoir for benthic organisms. Marine and coastal environments receive pharmaceuticals via river discharge and direct coastal WWTP outfalls, with studies documenting detectable concentrations of antibiotics and hormones in seawater and marine biota in multiple world regions (34). The concentration ranges encountered across these matrices span from sub-ng/L in drinking water and clean groundwater through ng/L in surface water and coastal environments to μg/L in WWTP influents and hospital effluents and mg/L at pharmaceutical manufacturing discharge points, imposing dramatically different demands on method sensitivity, selectivity, and preconcentration efficiency (35).
Green Analytical Chemistry (GAC): Concepts and Assessment of Greenness
Green Analytical Chemistry formally emerged from the application of the 12 Principles of Green Chemistry to analytical workflows, with Namiesnik and colleagues providing foundational definitions in the early 2000s, subsequently codified through the SIGNIFICANCE mnemonic (36). The core GAC principles emphasise direct analysis without sample preparation where possible; minimisation of sample size and number; use of in-line rather than off-line procedures; miniaturisation of instrumentation; elimination or substitution of hazardous reagents; reduction of energy consumption; and generation of as little waste as possible, with any waste being as non-hazardous as feasible (37). In pharmaceutical water analysis, these principles translate concretely into preference for miniaturised extractions over large-volume SPE, solvent substitution from halogenated organic solvents to bio-based or aqueous alternatives, and adoption of UHPLC over conventional HPLC (38).
However, a fundamental tension exists within GAC as applied to trace pharmaceutical monitoring that must be honestly acknowledged. Greener methods frequently sacrifice sensitivity, robustness, or analytical scope. Pharmaceuticals at ng/L concentrations in complex environmental matrices require aggressive preconcentration, high chromatographic efficiency, and sensitive mass spectrometric detection, all of which carry an environmental cost. A method that is analytically incapable of detecting carbamazepine or diclofenac at environmentally relevant concentrations is not useful regardless of its greenness score. Analytical performance is therefore the primary constraint, and sustainability improvements must be pursued within its bounds rather than at the expense of data quality (39).
To provide objective, quantitative assessment of analytical method greenness, several complementary tools have been developed. The Analytical Eco-Scale, introduced by Galuszka et al. in 2012, assigns penalty points to a method based on hazardous reagent volumes and nature, energy consumption, waste generation, and occupational hazard, scoring from 100 (ideal green) downward; scores above 75 are classified as excellent green (40). While operationally straightforward, the Eco-Scale is reductive: it does not account for method scope, sample throughput, or analytical performance, and can award high greenness scores to methods with poor detection limits or restricted compound coverage.
The Green Analytical Procedure Index (GAPI), developed by Plotka-Wasylka in 2018, addresses some of these limitations through a visual pentagram representation spanning the entire analytical procedure from sample collection through measurement and data processing, with each segment colour-coded to reflect environmental impact (41). GAPI provides a more holistic workflow view and has been widely adopted in the analytical chemistry literature. Its primary limitation is subjectivity in colour assignment and the absence of a single numerical comparator, complicating inter-method ranking. The AGREE (Analytical GREEnness) metric, published by Pena-Pereira et al. in 2020, represents the most recent and most comprehensive tool, integrating all 12 GAC principles into a single score from 0 (not green) to 1 (perfectly green) using a defined algorithm that accounts for reagent hazard, energy use, waste generation, and automation level (42). AGREE currently offers the most defensible and standardised greenness assessment and should be the default reporting metric in pharmaceutical water analysis publications. White Analytical Chemistry, proposed by Nowak et al., extends the framework further to encompass functionality, economic viability, and social acceptability alongside environmental impact, providing the broadest multi-criteria evaluation perspective (43). Collectively, these tools enable transparent greenness benchmarking alongside analytical performance data, a practice that should become standard in the field (44).
Sampling and Preservation with Reduced Environmental Footprint
Representative sampling is the foundation of reliable pharmaceutical monitoring and itself carries an environmental footprint that is frequently overlooked in the green analytical chemistry literature. Grab samples collected at a single time point provide instantaneous concentration data and are adequate for capturing acute contamination events or compliance spot checks, but are highly susceptible to temporal variability in pharmaceutical discharge and dilution, limiting their representativeness for characterising chronic exposure conditions (45). Composite samples, assembled by pooling discrete aliquots over defined time intervals using automated refrigerated samplers, provide a more integrated representation of mean pharmaceutical concentrations but require electrical power for refrigeration and pumping, generate cooling waste, and demand cold-chain logistics for transport to the laboratory, all of which increase the environmental footprint and operational cost of sampling campaigns (46).
Passive sampling has emerged as the most genuinely green sampling strategy for pharmaceutical monitoring, offering time-integrated concentration data over deployment periods of days to weeks without the need for pumps, power supply, or continuous operator attendance (47). The polar organic chemical integrative sampler (POCIS) is the most widely used device for hydrophilic pharmaceuticals, employing a polymeric sorbent (HLB or mixed-mode material) sandwiched between microporous polyethersulphone membranes that control diffusive uptake from the water column (48). Chemcatcher devices equipped with C18 or SDB-RPS discs serve a similar function for moderately hydrophobic compounds, while silicone-based low-density polyethylene (LDPE) sheets are used for hydrophobic analytes. Passive samplers have been successfully deployed in rivers, coastal waters, groundwater, and WWTP effluents for multi-class pharmaceutical monitoring, demonstrating detection of compounds at concentrations below the limits of conventional grab sampling methods (49).
The principal analytical challenge with passive samplers is conversion of the accumulated analyte mass to a time-weighted average concentration (TWAC) in water, which requires knowledge of the compound-specific sampling rate (Rs). Sampling rates are influenced by temperature, current velocity, biofouling of membrane surfaces, and the physicochemical properties of the analyte, introducing uncertainty into TWAC calculations (50). Performance reference compounds (PRCs)—compounds pre-loaded onto the sampler sorbent that dissipate at rates proportional to uptake—can correct for in situ hydrodynamic and temperature variability and substantially reduce TWAC uncertainty when properly selected and validated (51). Despite these complexities, passive samplers represent the most resource-efficient option for long-term surveillance monitoring of pharmaceuticals in rivers, coastal waters, and groundwater, and their integration into national monitoring frameworks under the EU Water Framework Directive has been actively explored (52).
Sample preservation practices carry their own environmental implications. Standard protocols employ sodium azide as a biocide to suppress microbial pharmaceutical degradation, ascorbic acid to quench chlorine residuals, and acidification to inhibit photolysis and hydrolysis during transport. Where analytically permissible, reducing preservative volumes, processing samples rapidly in the field, using amber glass rather than clear plastic containers to minimise photodegradation, and maintaining cold chain only where compound stability demands it can meaningfully reduce the material footprint of sampling operations (53). Field blanks, equipment blanks, and trip blanks must be consistently incorporated to discriminate genuine environmental contamination from procedural artefacts, which is particularly critical at ng/L detection levels where ambient laboratory contamination from ubiquitous pharmaceuticals such as ibuprofen and caffeine can be significant (54). Replacing single-use plasticware with reusable glass or stainless-steel sampling vessels further reduces the indirect environmental burden of monitoring programmes (45).
Green Sample Preparation and Preconcentration
Sample preparation is the most solvent-intensive and environmentally burdensome step in pharmaceutical analysis of environmental waters, accounting for the majority of organic waste generated and a substantial fraction of analysis time and analyst exposure to hazardous chemicals. Conventional large-volume SPE using 500 mL to 1 L water samples requires 15-30 mL of organic solvent for cartridge conditioning, washing, and elution per sample, generating several litres of hazardous waste per analytical batch. Green sample preparation therefore represents the highest-leverage target for reducing the environmental footprint of pharmaceutical water analysis (55). The following subsections critically evaluate the principal approaches.
Miniaturised and Solvent-Free Microextraction
Solid-phase microextraction (SPME), introduced by Pawliszyn and colleagues in the early 1990s, is the archetypal solvent-free extraction technique for environmental analysis (56). In SPME, analytes partition between the aqueous sample and a thin polymer or mixed-mode sorbent coating on a fused silica fibre, arrow-shaped device, or thin film, driven by the analyte distribution coefficient between the phases. Thermal desorption into a GC injector or brief solvent desorption for LC-MS/MS analysis eliminates organic solvent use entirely in the extraction step (57). Fibre SPME in direct immersion (DI-SPME) mode has been applied to NSAIDs, antibiotics, hormones, and antidepressants in surface water and wastewater, with polydimethylsiloxane-divinylbenzene (PDMS-DVB) and mixed-mode C18-SCX coatings providing broad pharmaceutical coverage (58). Thin-film microextraction (TFME) substantially increases the extraction phase volume-to-sample volume ratio compared to conventional fibres, improving enrichment factors and lowering method detection limits for polar pharmaceuticals including metformin, atenolol, and sulfamethoxazole in ng/L-spiked surface water matrices (59).
Stir-bar sorptive extraction (SBSE), marketed as Twister, employs a glass-coated magnetic stir bar bearing a PDMS or mixed-mode sorbent layer that concentrates analytes during magnetic stirring of the sample (60). Extraction phase volumes of 50-300 μL, much larger than fibre SPME, yield enrichment factors of 100-1000-fold for moderately hydrophobic pharmaceuticals (log Kow 2-4), making SBSE particularly effective for lipid regulators, antidepressants, and steroid hormones in surface water at low ng/L concentrations (61). Thermal desorption of SBSE bars is the primary desorption route for GC-MS analysis, while back-extraction into small organic solvent volumes followed by LC-MS/MS suits polar pharmaceutical targets. A shared limitation of SPME and SBSE is reduced efficiency for highly polar, ionised pharmaceuticals (log Kow < 1) such as metformin, methotrexate, and aminoglycosides; mixed-mode ion-exchange coatings partially address this but do not yet provide universal pharmaceutical coverage comparable to polymeric SPE sorbents (62).
Liquid-Phase Microextraction (LPME) Families
Dispersive liquid-liquid microextraction (DLLME), first described by Rezaee et al. in 2006, achieves high enrichment factors through rapid dispersion of a small extraction solvent volume into the aqueous sample using a water-miscible disperser solvent, forming a fine cloudy emulsion that maximises analyte mass transfer to the extraction phase (63). Centrifugation sediments the denser extraction solvent (typically 10-50 μL), which is collected by microsyringe for direct injection or evaporation and reconstitution. Conventional DLLME employed tetrachloroethylene or chlorobenzene as extraction solvents; greener variants substitute 1-dodecanol, undecanol, ethyl acetate, or hexyl acetate as low-toxicity alternatives, while using ethanol, methanol, or acetone as disperser solvents, substantially reducing the overall hazard profile of the procedure (64). Salting-out effects from sodium chloride addition enhance phase separation and extraction efficiency for polar pharmaceuticals and are particularly relevant for saline coastal or estuarine matrices.
Ultrasound-assisted DLLME and vortex-assisted DLLME variants replace the disperser solvent with physical energy input to generate the emulsion, further reducing organic reagent use and improving reproducibility (65). Air-assisted LLME, in which the two-phase system is repeatedly aspirated and dispensed through a syringe to achieve dispersion, eliminates the need for a disperser solvent entirely and is among the greenest LPME variants applicable to pharmaceutical analysis in water (66). Hollow-fibre liquid-phase microextraction (HF-LPME) immobilises the extraction solvent within the pores of a porous polypropylene hollow fibre, providing a supported liquid membrane through which analytes diffuse from the donor aqueous phase to an acceptor phase inside the fibre lumen (67). HF-LPME offers high selectivity and enrichment factors of several hundred-fold for pharmaceuticals, but requires 30-60 min extraction times, is fragile and difficult to automate, and provides limited throughput, confining its utility to targeted research applications rather than high-volume routine monitoring.
Greener SPE Approaches
Despite the appeal of solvent-free microextraction, SPE remains the dominant preconcentration technique for pharmaceutical monitoring in environmental waters, owing to its validated multi-residue performance across diverse compound classes and matrices, regulatory acceptance, and compatibility with large sample volumes needed for ng/L sensitivity in drinking water (68). Green SPE practice focuses on minimising solvent volumes, developing reusable formats, and employing novel sorbents with superior selectivity that reduce washing and conditioning requirements. Polymeric reversed-phase sorbents, particularly Oasis HLB (hydrophilic-lipophilic balance) copolymers, have become the standard choice for multi-class pharmaceutical SPE due to their broad polarity range retention and compatibility with aqueous sample loading without sorbent desiccation (69). Optimised HLB-based protocols using 200 mg cartridges and 2 × 2 mL methanol elution achieve quantitative recoveries for over 100 pharmaceutical compounds from 200 mL water samples with total organic solvent use below 8 mL per sample (70).
Online SPE directly coupled to the LC-MS/MS system via a switching valve eliminates the off-line evaporation and reconstitution steps required in conventional SPE, reducing solvent consumption by 50-70%, minimising analyte losses from evaporation, and substantially improving throughput and reproducibility through automation (71). Turbulent flow chromatography (TFC) and restricted-access material (RAM) columns extend this approach to direct injection of urine, plasma, and high-turbidity environmental samples with online removal of matrix components prior to analytical separation (72). Molecularly imprinted polymers (MIPs) synthesised against target pharmaceutical templates provide exquisitely selective sorbents that retain analytes from complex matrices with minimal co-extraction of interfering compounds, reducing cleanup solvent requirements and improving selectivity in HRMS non-target workflows (73). Metal-organic frameworks (MOFs), including ZIF-8, MIL-53, and UiO-66 variants, offer exceptional surface areas (1000-3000 m2/g) and tunable pore chemistry enabling simultaneous retention of structurally diverse pharmaceuticals from water in gram-scale sorbent formats (74). Magnetic dispersive SPE (MSPE) using Fe3O4 nanoparticles functionalised with mixed-mode sorbent coatings allows rapid sorbent-analyte contact at high surface area and magnetic recovery without centrifuge columns or cartridges, reducing solvent use and analysis time for multi-class pharmaceutical extraction from surface water (75). However, a rigorous lifecycle perspective is essential when evaluating these novel sorbents: energy-intensive synthesis routes, hazardous precursors, unknown aquatic ecotoxicity, and undocumented end-of-life disposal pathways mean that net environmental benefit over established polymeric sorbents remains unproven and should not be assumed (76).
Alternative Solvents and Reagents
Solvent substitution is a cornerstone GAC strategy, and bio-based solvents derived from renewable feedstocks have attracted substantial research interest as replacements for conventional polar aprotic and halogenated solvents in pharmaceutical extraction. 2-Methyltetrahydrofuran (2-MeTHF), derived from agricultural waste furfural, offers polarity and miscibility properties similar to diethyl ether with improved biodegradability and a bio-renewable origin, and has demonstrated acceptable recovery for NSAIDs and antibiotics in DLLME applications (77). Ethyl lactate, cyrene (derived from cellulose), and limonene represent additional bio-based candidates with reduced acute toxicity and improved environmental profiles relative to acetonitrile or dichloromethane (78). However, bio-based designation does not automatically confer environmental safety: solvent recovery efficiency in laboratory waste streams, biodegradation rates in aquatic receiving environments, and interactions with co-contaminants require case-by-case evaluation before green claims can be substantiated.
Ionic liquids (ILs) have been extensively investigated as green extraction media for pharmaceutical analysis due to their negligible vapour pressure, thermal stability, and the capacity to tune physicochemical properties through cation-anion pairing (79). Imidazolium, ammonium, and phosphonium-based ILs have been applied in DLLME and in situ IL-DLLME formats for NSAIDs, antibiotics, and hormones in water, often yielding enrichment factors exceeding 1000-fold. However, the green credentials of ILs require critical scrutiny: many exhibit significant and persistent aquatic toxicity, particularly towards algae and invertebrates; their synthesis routes frequently involve hazardous halogenated reagents; and biodegradation is limited for most commercially available variants (80). Deep eutectic solvents (DES), formed by mixing a hydrogen bond acceptor (typically choline chloride) with a hydrogen bond donor (urea, glycerol, citric acid, or phenolic acids) in defined molar ratios, present a substantially more credible green profile: most DES components are food-grade, non-toxic, and biodegradable, and DES preparation requires no synthesis, merely mixing at mild temperatures (81). Hydrophobic DES (HDES), prepared from combinations of fatty acids, menthol, or thymol, are immiscible with water and have been applied as extraction phases in DES-based DLLME for hydrophobic pharmaceuticals including diclofenac, ibuprofen, and hormones from surface water with competitive enrichment factors (82). Principal practical limitations of DES include high viscosity at room temperature, which complicates injection and pipetting, restricted analyte polarity range depending on DES composition, and incomplete toxicological characterisation of DES mixtures and their degradation products in environmental matrices (83).
Direct Analysis and Minimal Preparation Approaches
For water matrices with pharmaceutical concentrations in the μg/L range-WWTP influent, hospital effluents, and pharmaceutical manufacturing discharges-dilute-and-shoot (DaS) approaches requiring only filtration through 0.2 μm membrane filters and a defined dilution factor prior to direct LC-MS/MS injection represent the simplest and greenest sample preparation option (84). DaS eliminates organic solvent use in sample preparation entirely, reduces analysis time to 30-60 minutes per sample including chromatography, and avoids the analyte losses and matrix enrichment artefacts associated with extraction procedures. The approach requires careful management of matrix effects through matrix-matched calibration and stable isotope-labelled internal standards, as dissolved organic matter and ionic components of WWTP effluents can substantially suppress or enhance electrospray ionisation signals (85). For samples where matrix effects are poorly reproducible, online SPE provides automated dilution and partial matrix removal within a single analytical run, combining the solvent efficiency of DaS with improved matrix tolerance for μg/L-range samples (86).
QuEChERS (Quick, Easy, Cheap, Effective, Rugged, Safe)-inspired approaches, originally developed for pesticide analysis in food matrices, have been adapted for pharmaceutical extraction from sediment, sludge, and biota matrices, providing rapid acetonitrile-based extraction followed by dispersive SPE cleanup using primary-secondary amine (PSA), C18, and graphitised carbon sorbents in a single 15-minute workflow with minimal solvent use (87). The approach achieves acceptable recoveries for a broad pharmaceutical spectrum from solid matrices with total solvent consumption below 20 mL per sample, representing a substantial green improvement over conventional pressurised liquid extraction-SPE sequences (88).
Green(er) Separation and Instrumental Detection
Liquid Chromatography Improvements
The transition from conventional HPLC (5 μm particle columns, 1-2 mL/min flow rates, 30-60 min run times) to UHPLC operating with sub-2-μm particles at pressures above 600 bar has delivered some of the most impactful practical green improvements in pharmaceutical water analysis (89). Analytical run times are reduced 3-5-fold; mobile phase consumption decreases proportionally; improved peak efficiency enables detection of lower-abundance compounds from smaller injection volumes; and higher sensitivity per unit time allows shorter sample preconcentration. A typical 10-minute UHPLC method consuming 4 mL of mobile phase per run compared to a 40-minute HPLC method consuming 80 mL represents a 95% reduction in per-sample solvent use for separation alone (90). Core-shell particle columns, combining a solid silica core with a porous shell, achieve near-UHPLC efficiency at conventional HPLC pressures (< 400 bar), providing a cost-effective greenness improvement route for laboratories without UHPLC infrastructure (91).
The choice of organic modifier in the mobile phase carries environmental implications: methanol, derived from natural gas or bio-based routes, has substantially lower acute toxicity and better biodegradability than acetonitrile, whose production involves hydrogen cyanide as a precursor (92). For pharmaceuticals amenable to reversed-phase separation with either modifier, substitution of acetonitrile with methanol reduces both the hazard of mobile phase waste and chromatographic system environmental burden, though slightly different selectivity may necessitate gradient re-optimisation. Microflow LC operating at 10-100 μL/min further reduces solvent consumption by 10-100-fold compared to UHPLC and improves ionisation efficiency in ESI-MS through reduced ion suppression at lower flow rates, but operational complexity, susceptibility to particulate blockage, and long equilibration times limit its suitability for routine high-throughput environmental monitoring (93).
Alternative Separations
Capillary electrophoresis (CE) is intrinsically among the greenest analytical separation techniques, consuming only nanolitres of buffer solution per injection with virtually no organic waste generation (94). CE modes including capillary zone electrophoresis (CZE), micellar electrokinetic chromatography (MEKC), and capillary electrochromatography (CEC) have been applied to pharmaceutical residues in water samples, with MEKC using surfactant-modified buffers providing acceptable separation of structurally similar pharmaceutical classes such as sulphonamide antibiotics and beta-blockers (95). The technique offers high efficiency, low matrix injection volumes, and rapid method development for ionisable pharmaceuticals. However, the inherently short detection path length of capillary formats limits UV sensitivity and coupling to MS detection via sheath-flow ESI or sheathless interfaces introduces technical complexity that has constrained the widespread adoption of CE-MS in environmental pharmaceutical monitoring (96). CE therefore currently occupies a specialised niche for targeted analysis of ionisable pharmaceutical classes in cleaner matrices rather than serving as a general-purpose replacement for LC-MS.
Supercritical fluid chromatography (SFC) using CO2-based mobile phases supplemented with polar modifiers offers a genuinely green separation alternative for moderately hydrophobic pharmaceuticals, with CO2 as the primary mobile phase component being non-toxic, non-flammable, and readily recycled within closed-loop instrument configurations (97). SFC has demonstrated efficient separation of lipid-soluble vitamins, steroids, and some antibiotic classes with substantially reduced organic modifier consumption compared to RPLC, and is gaining traction in pharmaceutical quality control applications. Its applicability to the polar, ionisable pharmaceutical classes most frequently detected in aquatic environments remains more limited, representing an important development priority.
Detection Strategies
Tandem quadrupole mass spectrometry in multiple reaction monitoring (MRM) mode, coupled to UHPLC via positive and negative electrospray ionisation with polarity switching, is the established analytical workhorse for targeted quantitative pharmaceutical monitoring in environmental water samples (98). The combination of precursor ion selection, collision-induced dissociation to compound-specific product ions, and product ion detection provides unmatched selectivity, effectively eliminating co-eluting matrix interferences that would compromise UV or single-quadrupole MS detection. LOQs in the 1-10 ng/L range in surface water and 0.1-1 ng/L in groundwater are routinely achievable with 200-500 mL SPE preconcentration, covering regulatory priority compounds including diclofenac (EU WFD Environmental Quality Standard: 100 ng/L), 17β-estradiol (0.4 ng/L EQS), and 17α-ethinylestradiol (0.035 ng/L EQS) (99). Simultaneous positive/negative polarity switching in a single LC-MS/MS run enables co-analysis of acids (NSAIDs, fibrates), bases (antibiotics, antidepressants), and neutral compounds (carbamazepine) without the need for separate injections, maximising per-injection analytical efficiency (100).
High-resolution mass spectrometry (HRMS) using Orbitrap or quadrupole time-of-flight (QTOF) instruments has transformed pharmaceutical environmental monitoring by enabling suspect and non-target screening alongside targeted quantification in a single analytical run (101). Suspect screening queries accurate mass features detected in the chromatogram against databases of known pharmaceuticals, their metabolites, and transformation products, enabling retrospective identification of compounds excluded from targeted methods without additional sample analysis (102). Non-target screening generates a comprehensive mass spectral inventory of all detected features, which can be interrogated for unknown transformation products, novel pharmaceutical residues, or previously unrecognised environmental contaminants (103). HRMS data processing workflows combining automated feature extraction, database matching, and fragmentation-based structural elucidation have become increasingly tractable through dedicated software platforms such as MZmine, XCMS, Compound Discoverer, and MS-DIAL (104). The primary limitations of HRMS relative to MRM-based MS/MS are lower sensitivity for targeted quantification, higher instrument acquisition and operating costs, greater data storage and processing demands, and the need for specialist expertise in non-target data interpretation (105). The optimal strategy for most monitoring programmes is a tiered approach: routine UHPLC-MS/MS for targeted priority compound quantification combined with periodic HRMS campaigns for surveillance, suspect screening, and non-target discovery.
Reducing Instrument Footprint
Mass spectrometers and their associated high-vacuum turbopumps, rotary backing pumps, ion sources, and data systems are substantial consumers of electrical energy, nitrogen or helium gas for drying, desolvation, and collision, and water for heat exchange, creating an instrument-level environmental footprint that receives insufficient attention relative to sample preparation greening (106). Practical measures to reduce this footprint include: scheduling analytical batches to consolidate instrument operating time and minimise standby heating cycles; using nitrogen generators from compressed air rather than cylinder gas, eliminating transport emissions from high-pressure gas logistics; implementing solvent recycling for mobile phase waste streams where chemical compatibility permits; adopting shorter analytical columns (50 mm rather than 100-150 mm) to reduce per-run solvent use while maintaining required resolution; and implementing predictive maintenance protocols that reduce system failures, column replacements, and associated re-analysis requirements (107). Emerging miniaturised mass spectrometry platforms including portable ion trap and miniature magnetic sector instruments offer potential for field-deployable pharmaceutical monitoring with substantially reduced power requirements, though currently at significantly higher LOQs than laboratory HRMS systems (108).
Method Validation and QA/QC for Environmental Monitoring
Rigorous method validation is an indispensable prerequisite for any analytical procedure deployed for pharmaceutical monitoring in aquatic environments, establishing the fitness-for-purpose of the method under the specific matrix and concentration conditions of its intended application. Standard validation parameters encompass recovery (mean extraction efficiency across the concentration range and matrices of interest), matrix effects (percentage signal suppression or enhancement in ESI-MS relative to pure solvent standards), linearity and working range of calibration, limits of detection (LOD, signal-to-noise ratio 3:1 or equivalent blank-based calculation) and quantification (LOQ, signal-to-noise ratio 10:1), within-day repeatability (intra-day RSD), between-day intermediate precision (inter-day RSD), and trueness assessed against certified reference materials or spiked procedural blanks (109). The ISO/IEC 17025 standard and SANCO/SANTE guidelines for pesticide residue analysis provide internationally recognised validation frameworks applicable by analogy to pharmaceutical environmental analysis (110).
Matrix effects are the most analytically critical validation parameter for LC-MS/MS-based pharmaceutical monitoring and represent the primary source of quantification bias in environmental samples. Dissolved organic carbon, humic and fulvic acids, salts, co-extracted endogenous compounds, and surfactants from WWTP effluents can suppress electrospray ionisation by 20-90% for certain pharmaceutical-matrix combinations, or in some cases enhance ionisation, leading to systematic under- or over-estimation of true concentrations (111). Matrix-matched calibration prepared in analyte-free matrix extracts processed through the complete sample preparation procedure is the minimum acceptable correction approach; isotopically labelled internal standards (2H, 13C, or 15N analogues of target analytes) provide superior correction by tracking matrix effects and recovery losses simultaneously throughout the analytical workflow (112). When isotopically labelled standards are not available for all target compounds, structurally analogous surrogate standards with similar physicochemical properties can partially substitute, though with reduced compensation accuracy.
LOD and LOQ values should be reported in final sample concentration units (ng/L or μg/L in water) after accounting for the full sample preparation enrichment factor, not as instrument-level values that ignore preconcentration. Reporting instrument LOQ without correcting for recovery losses and preconcentration factor systematically underestimates true method LOQ and misrepresents method capability to end users and regulators (113). Comprehensive blank analysis incorporating reagent blanks, procedural blanks processed through the complete sample preparation sequence, and field blanks collected, transported, and processed alongside real samples is essential to identify and quantify contamination at each analytical stage, particularly important at ng/L working concentrations where background pharmaceutical contamination of laboratory reagents, glassware, and equipment can be significant (114). Carryover assessment is mandatory when high-concentration WWTP or manufacturing effluent samples are analysed in the same sequence as low-concentration drinking water or groundwater samples on shared instrumentation.
Inter-laboratory method comparability remains a persistent challenge in pharmaceutical water monitoring, with proficiency testing schemes and ring trials demonstrating coefficient of variation values of 20-50% or more for reported concentrations of the same pharmaceutical compound in blind split samples across different laboratories (115). Harmonisation of extraction procedures, analytical platforms, calibration approaches, and data reporting conventions is essential for generating the consistent, comparable datasets needed to evaluate temporal and spatial contamination trends across monitoring networks. Active participation in proficiency testing programmes, adoption of reference methods developed through interlaboratory validation, and use of certified reference materials where available (e.g., NIST SRM 1950 for plasma, ERM-CA713 for wastewater) contribute to inter-laboratory standardisation (116). Critically, greenness assessment results and their supporting data should be reported alongside full validation tables in methods publications, enabling both the analytical performance and the environmental sustainability of proposed methods to be assessed by readers and compared across the literature.
Comparative Assessment and Decision Framework
The selection of an appropriate analytical workflow for pharmaceutical monitoring requires simultaneous, multi-criteria evaluation of analytical performance (sensitivity, selectivity, multi-residue scope), greenness (solvent use, waste generation, energy consumption, operator hazard), practicality (throughput, automation, equipment requirements, analyst skill), and cost. No single method excels across all dimensions in all matrices, and an honest decision framework must acknowledge trade-offs rather than endorsing a single universally optimal approach (117).
For high-throughput WWTP surveillance monitoring where pharmaceutical concentrations are in the μg/L range and large sample numbers must be processed efficiently, online SPE-UHPLC-MS/MS with DaS preparation for pre-treated effluents represents the most pragmatically green option. Automation eliminates solvent-intensive off-line extraction, batch scheduling maximises instrument utilisation, and polarity-switching MRM methods enable simultaneous coverage of 50-100+ pharmaceutical targets per injection. AGREE scores of 0.55-0.70 are achievable for well-optimised online SPE methods, with the primary greenness limitations arising from mobile phase consumption during chromatographic separation and mass spectrometer energy use rather than sample preparation (118).
Ultra-trace drinking water analysis targeting sub-ng/L concentrations of priority pharmaceuticals including ethinylestradiol (EU EQS: 0.035 ng/L) necessitates large-volume SPE (500 mL-1 L) with optimised HLB sorbents and low elution volumes to achieve required sensitivity. In this scenario, substituting methanol for acetonitrile as both mobile phase modifier and SPE elution solvent, minimising conditioning and washing solvent volumes through HLB sorbent selection, and implementing online evaporation-free reconstitution represent achievable greenness improvements while maintaining LOQs at 0.1-1 ng/L. SPME and DLLME are generally inadequate for drinking water pharmaceutical analysis at regulatory EQS levels and should not be selected on greenness grounds if analytical targets cannot be met (119).
High-salinity coastal and seawater matrices present specific challenges: salt-induced ion suppression in ESI, phase separation interference in DLLME, and reduced SPE recoveries for ionised pharmaceuticals due to competition from seawater cations. RAM sorbents and HLB-based SPE with enhanced aqueous washing steps, or salting-out assisted DLLME optimised for specific ionic strength ranges, are more robust than conventional fibre SPME or HF-LPME in marine matrices (120). Passive sampling with POCIS and Chemcatcher is particularly well-suited to coastal pharmaceutical monitoring, providing time-integrated exposure data without the logistical complexity of large-volume water collection at sea, and enabling detection of episodic contamination events from seasonal WWTP or river discharge that grab sampling campaigns would miss (47).
Sediment and sludge analysis requires solvent-based extraction given the strong sorption of hydrophobic pharmaceuticals, hormones, and fluoroquinolone antibiotics to organic matter and mineral surfaces, making solvent-free microextraction approaches inapplicable (121). Pressurised liquid extraction (PLE/ASE) with methanol-water or acetonitrile-water mixtures, combined with dispersive SPE cleanup in a QuEChERS-inspired format, represents the most green-performant option, reducing solvent consumption per sample to below 20 mL compared to 50-100 mL for conventional Soxhlet or sonication-followed-by-column-SPE sequences (88). Novel MOF and MIP sorbent claims for solid matrix applications require lifecycle scrutiny before green credentials can be accepted, as synthesis energy costs, precursor hazard, and disposal implications are rarely evaluated in publications presenting these materials (76).
Knowledge Gaps and Future Directions
Despite the substantial progress reviewed above, several critical knowledge gaps and development priorities remain that constrain the scientific community's ability to monitor pharmaceutical contamination in aquatic environments in a genuinely sustainable manner. These are outlined below with specific recommendations for research investment.
Standardised greenness reporting is inconsistently applied and inconsistently demanded in the pharmaceutical water analysis literature. A survey of publications describing new analytical methods for pharmaceutical environmental monitoring reveals that fewer than 30% report any formal greenness assessment, and among those that do, there is minimal consistency in the choice of metric, the parameters included, and the level of supporting data provided (122). Journals publishing in environmental analytical chemistry should mandate reporting of at least one validated greenness metric-preferably AGREE-alongside full validation performance tables as a condition of publication for methods papers, analogous to the established requirement for reporting LOD, LOQ, and recovery data. Without this standardisation, meaningful inter-method greenness comparison is impossible and claims of "green" methodology remain unverifiable.
Hybrid workflows combining passive sampling with minimal-solvent laboratory analysis represent one of the most promising development frontiers. Passive samplers already substantially reduce the field footprint of monitoring campaigns; coupling their use with on-sorbent thermal desorption into GC-MS or LC-MS without liquid elution, or with in situ SPME approaches for volatile and semi-volatile pharmaceutical transformation products, would enable near-solvent-free end-to-end workflows (123). Proof-of-concept demonstrations exist for selected pharmaceutical classes, but systematic multi-class validation across diverse aquatic matrices and environmental conditions is needed before operational deployment in regulatory monitoring programmes.
Lifecycle assessment of novel extraction materials represents perhaps the most significant unaddressed gap in the green sample preparation literature. Publications on MOF, MIP, magnetic nanoparticle, or DES-based extraction systems routinely report reduced organic solvent use as evidence of greenness without providing synthesis energy costs, precursor toxicity profiles, device reusability over realistic analytical cycles, or end-of-life fate and disposal pathways (76, 124). A standardised lifecycle assessment (LCA) framework specifically designed for analytical microextraction devices-covering raw material extraction, synthesis, use phase, and disposal-would enable objective comparison of novel and conventional sorbents. Until such frameworks are developed and applied, claims of net environmental superiority for novel sorbents over established polymeric phases should be treated with caution.
Portable and field-deployable analytical platforms offer transformative potential for reducing the transport, storage, cold-chain, and laboratory infrastructure burden of pharmaceutical monitoring, particularly in low- and middle-income countries where centralised laboratory access is constrained. Miniaturised ion trap mass spectrometers, paper-based electrochemical aptasensors, molecularly imprinted polymer-coated quartz crystal microbalance sensors, and SERS-active substrates have each demonstrated ng/L sensitivity for selected pharmaceuticals under controlled conditions (108, 125). However, robust performance in field-realistic conditions-with variable temperature, humidity, turbidity, ionic strength, and complex co-contaminant profiles-remains to be demonstrated for most platforms. An envisaged tiered field-laboratory workflow, in which portable sensors perform rapid on-site screening with laboratory UHPLC-MS/MS confirmation of presumptive positives only, would reduce the volume of samples requiring full analytical processing by potentially 80-90%, substantially improving the sustainability of large-scale monitoring programmes while maintaining the data quality needed for regulatory decision-making (126).
CONCLUSIONS
This review has critically examined the current state and future potential of green analytical approaches for the detection of pharmaceutical contaminants in aquatic environments, spanning sampling, sample preparation, chromatographic separation, mass spectrometric detection, method validation, and greenness assessment. The following conclusions are offered as practically actionable guidance for researchers and monitoring laboratories.
Genuine greenness in pharmaceutical water analysis is achievable without unacceptable compromise of analytical performance when method selection simultaneously applies both sustainability and fitness-for-purpose criteria. UHPLC-MS/MS with online SPE, or offline miniaturised SPE using optimised HLB protocols with minimal solvent volumes, currently represents the best-performing intersection of analytical capability and environmental responsibility for most aquatic matrices. The transition from conventional HPLC to UHPLC alone-achievable without additional capital investment beyond column and method re-optimisation-delivers 70-90% reduction in per-sample mobile phase consumption and represents the single highest-impact green improvement available to most analytical laboratories. Microextraction approaches including SPME, thin-film SPME, and greener DLLME variants are valuable for targeted applications in cleaner or moderately complex matrices, but must not be selected on greenness grounds alone if they cannot reliably detect target pharmaceuticals at environmentally and regulatorily relevant concentrations.
Greenness assessment using AGREE, reported alongside full validation data, should become a mandatory element of methods publications in pharmaceutical environmental monitoring, enabling transparent comparison and progressive improvement of the sustainability profile of analytical approaches. Passive sampling deserves substantially broader integration into routine monitoring frameworks than it currently receives, offering superior temporal representativeness and a dramatically lower field footprint compared to conventional grab or composite sampling without significant compromise of concentration data quality when PRC-based calibration is applied. Novel sorbents-MOFs, MIPs, MSPE, DES extraction phases-should be subjected to lifecycle assessment before their green credentials are accepted; the analytical community has a responsibility to apply the same critical scrutiny to the environmental impact of its own materials and tools that it applies to the environmental contaminants it measures. Future investment should prioritise standardised greenness reporting frameworks, hybrid passive-microextraction-LC-MS workflows, lifecycle assessment of novel analytical materials, and robust validation of portable field-deployable screening platforms. The analytical chemistry community has both the tools and the obligation to monitor pharmaceutical pollution more sustainably-a responsibility commensurate with the scale and urgency of the contamination it seeks to characterise.
REFERENCES


Jovin K Jose, Praseetha K, Green Analytical Approaches for Detection of Pharmaceutical Contaminants in Aquatic Environments: A Critical Review, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 542-565, https://doi.org/10.5281/zenodo.20508087
 10.5281/zenodo.20508087
10.5281/zenodo.20508087