
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Associate Professor, Pharmaceutical Chemistry, Department of Pharmacy, Govt. Medical College, Kottayam 686008
The imidazole scaffold is an important class of antifungal agents due to its ability to inhibit fungal sterol synthesis. Antifungal drugs may develop resistance, and research is crucial to developing new ones. In the present study, imidazole–5–ones and imidazole-4-ones were selected from a database and tested in silico using molecular docking, molecular dynamics, pharmacokinetics, and toxicity analyses to predict antifungal activity. Ketoconazole combined with a fungal target was used as the docking receptor. The AI-based Ginna Docker from Neurosnap AI was used, followed by the Alpha Flow dynamics simulator, ADMETAI, and e-tox drug toxicity assessment. The HITS selected were (5Z)-2-anilino-5-(1,3-benzodioxol-5-ylmethylidene)-3-methylimidazol-4-one(-8.18 kcal/mol) and (4Z)-4-[(4-methylphenyl)methylidene]-2-piperidin-1-yl-1H-imidazol-5-one(-7.64kcal/mol) against Ketoconazole (-9.43 Kcal/mol). The ADMET parameters were satisfactory, with toxicity scores ranging from 0.550 to 0.618. The highest confidence Model portion was selected for the molecular docking study. Out of six Imidazoles, the affinity ranged between -7.10 and 8.18 Kcal/mol. The present study gave an insight into imidazole compounds and their antifungal potential. Further in vivo and clinical studies after lead optimization are proposed.
Fungal infections are considered a global health challenge due to drug resistance and their severity. Patients on steroid drug treatment are prone to fungal infection; Candida, Aspergillus, and Cryptococcus are the major causes of morbidity and mortality. [1]
Azole anti-fungal drugs have broad-spectrum activity. Ketoconazole prevents the fungal ergosterol biosynthesis. Imidazole heterocyclic compounds possess antifungal, antiparasitic, antiviral, and anticancer activities. [2] The imidazole 4–ones and 5- ones can be structurally optimized to develop novel, effective antifungal agents. The carbonyl mediates, and the imidazole ring facilitates hydrogen bond formation andπ-πinteractions at enzyme active sites. [3] Molecular docking can predict the biological activity of compounds before biological testing. [4] In combination with ADMC and toxicity parameters, unwanted compounds can be eliminated from screening. [5]
Materials and Methods the present study was designed as an in silico molecular docking analysis to identify the antifungal potential of selected imidazole–4–ones and imidazole–5–ones. The study was based on Molecular docking using GINNA AI from Neurosnap.inc and Alpha-Flow, and ADMET AI, including a toxicity study. The workflow included Ligand and protein target optimization using the PDB optimizer tool, molecular docking, comparative analysis with ketoconazole, toxicity predictions, and interpretation. The three-dimensional structure of the antifungal target protein (2V0M) was used in a molecular docking study. [6] The available structural models were evaluated using the Local distance difference test (pLDDT), Structural uniqueness, and root-mean-square deviation (RMSD) prior to docking. The top-ranked model exhibited the highest confidence score (mean pLDDT: 81.404) and was selected as the receptor for all analyses. Water molecules, reducing ligands, and nonessential heteroatoms were removed prior to receptor preparation, and polar hydrogens and Kolman's charges were assigned according to standard protocols. [7]
Ligand Preparation and Molecular Docking
Three imidazole–4–ones and three imidazole-5-ones were selected as test ligands. Ketoconazole was included as a reference antifungal drug for comparative evaluation. Structures were optimized and converted to the required docking format. For docking, Ginna AI from neurosnap.inc was used. The receptor was prepared by repairing missing atoms, cleaning the geometry, and adding polar hydrogen atoms using Bio-via Discovery Studio Visualizer. The receptor was kept rigid, and ligand flexibility was allowed during docking calculations. Binding energy was estimated in kcal/mol, with negative values indicating strong binding. [3] Poses were ranked by calculated binding energy.
ADMET AI and Toxicity Prediction. The docked ligands were used again for ADMET analysis. ADMET AI from NeuroSnap AI replaced conventional algorithm-based calculations, yielding accurate values. [8] The toxicity parameters were calculated from the results of the E tox drug toxicity assessment platform powered by NeuroSnap AI. [9] The parameters obtained were used as supporting indicators to prioritize ligands and optimize them for further biological study. The data obtained were compared with those for Ketoconazole compounds, which were ranked by binding affinity, and descriptive statistics were used to summarize docking performance.
Results and Discussion:
2D diagram
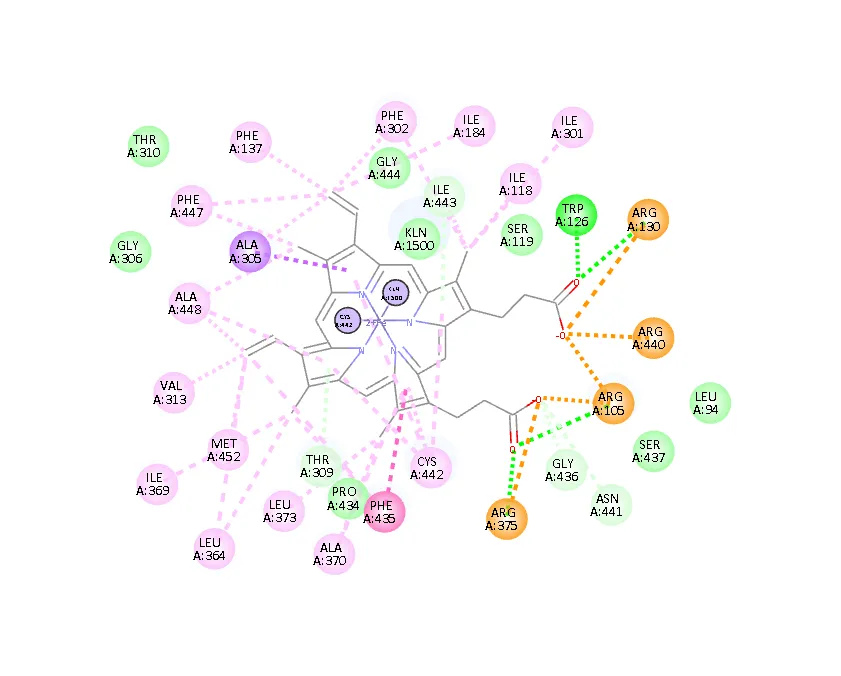
Figure 1: Pharmacophoric interaction sites of target 2V0

Figure 2: Crystal structure of human P450 3A4 in complex with ketoconazole
Table 1: Results of molecular docking of selected imidazole 5 and 4 - ones against fungal target 2V0M
|
Compound |
Binding Energy (kcal/mol) |
Rank |
Interpretation |
|
|
Ketoconazole |
-9.43 |
1 |
Reference |
|
|
CID 56676630 |
-8.18 |
2 |
Excellent |
|
|
CID 1354933935 |
-7.64 |
3 |
Very good |
|
|
CID 53308976 |
-7.43 |
4 |
Good |
|
|
CID 135398522 |
-7.39 |
5 |
Good |
|
|
CID 135493937 |
-7.18 |
6 |
Moderate |
|
|
CID 53309118 |
-7.10 |
7 |
Moderate |
|
|
Parameter |
Value |
|||
|
Number of compounds |
6 |
|||
|
Mean docking score (kcal/mol) |
-7.49 |
|||
|
Population SD |
0.36 |
|||
|
Minimum |
-8.18 |
|||
|
Maximum |
-7.10 |
|||
|
Range |
1.08 |
|||
Interpretation of Statistics
With a population standard deviation of 0.36 kcal/mol and a mean docking score of -7.49 kcal/mol, the six imidazole derivatives showed comparatively consistent projected binding affinities. Ketoconazole continued to be the greatest binder overall (-9.43 kcal/mol), while CID 56676630 was the best-performing derivative (-8.18 kcal/mol). Hypothesis tests (t-tests/ANOVAs) are not statistically acceptable because each drug in the dataset has a single docking score and there are no repeated docking runs or experimental observations. The manuscript should only include descriptive statistics.
Table 2: ADMET prediction of selected ligands
|
PubChem ID IUPAC name |
Log P |
Hydrogen bond _acceptors |
Hydrogen_ Bond _donors |
Lipinski |
QED |
BBB_Martins |
|
47576 Ketoconazole (reference) |
4.2058 |
7 |
0 |
3 |
0.4554 |
0.4644 |
|
53308976 (5Z)-5-(1,3-benzodioxol-5-ylmethylidene)-2-(4-hydroxyanilino)-3-methylimidazol-4-one |
2.4019 |
6 |
2 |
4 |
0.6494 |
0.5481 |
|
53309118 (5Z)-5-(1,3-benzodioxol-5-ylmethylidene)-2-(4-methoxyanilino)-3-methylimidazol-4-one |
2.7049 |
6 |
1 |
4 |
0.8611 |
0.7903 |
|
56676630 (5Z)-2-anilino-5-(1,3-benzodioxol-5-ylmethylidene)-3-methylimidazol-4-one |
2.6963 |
5 |
1 |
4 |
0.864 |
0.9482 |
|
135398522 (4Z)-2-anilino-4-(1,3-benzodioxol-5-ylmethylidene)-1H-imidazol-5-one |
2.3541 |
5 |
2 |
4 |
0.8355 |
0.9363 |
|
135493935 (4Z)-4-[(4-methylphenyl)methylidene]-2-piperidin-1-yl-1H-imidazol-5-one |
2.3076 |
3 |
1 |
4 |
0.7951 |
0.9407 |
|
135493937 (4Z)-4-[(4-chlorophenyl)methylidene]-2-piperidin-1-yl-1H-imidazol-5-one |
2.6526 |
3 |
1 |
4 |
0.808 |
0.9626 |
Table 3 Predicted Toxicity Profile of Screened Compounds
The table below summarizes the toxicity prediction results
|
CID |
Chemical structure |
Tox-score |
SAscore |
|
53308976 |
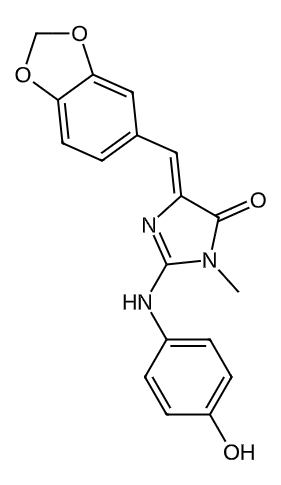
|
0.5558779640457661 |
0.2010315247097754 |
|
|
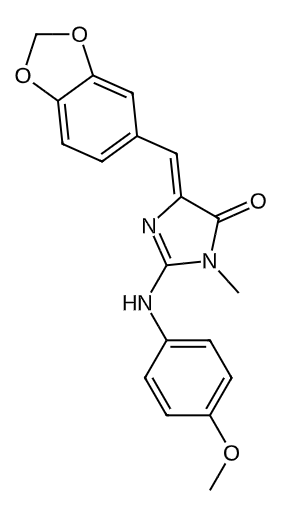
|
0.5431692972577149 |
0.2213766350205049 |
|
56676630 |
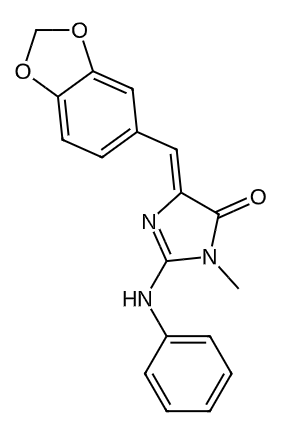
|
0.5531245082928624 |
0.2254174245347707 |
|
|
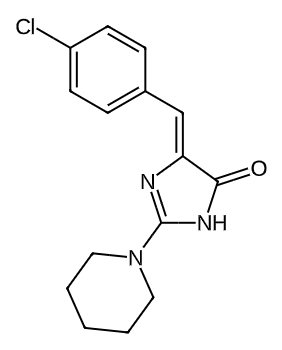
|
0.6186252907990432 |
0.2242816467390413 |
|
135398522 |
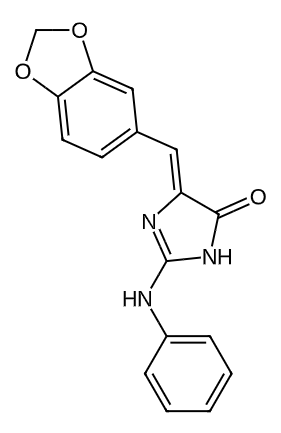
|
0.6097471044693151 |
0.2148496718620746 |
|
135398522 |
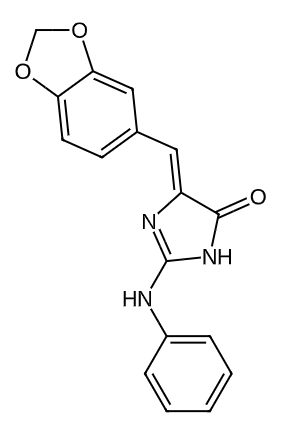
|
0.5240686880083768 |
0.087393271635275 |
|
Ketoconazole |
|
0.524069 |
0.087393 |
The structure compatibility assessment of the antifungal target protein (P.D.B9D: 2VOM) showed high confidence across the predicted models. The pLDDT values were evaluated, and a score of 81.404 was considered excellent stability. The PLDTT values of all ligands ranged from 64.5 to 77.90, confirming that the overall structural prediction was accurate and suitable for the study. [13] The reference model has an RMSD of 20-29 Å, confirming structural flexibility. [14] The uniform uniqueness scores indicated comparable structural Diversity among predicted models. The highest-ranked model was selected for receptor preparation. The molecular docking analysis of six imidazole derivatives against the selected antifungal target protein. Ketoconazole was used as a reference standard (-9.43 kcal/mol). The best HIT selected from PubChem ID 56676630 gave an excellent value (-8.18 Kcal/mol). The difference from the standard was only 1.25 Kcal/mol, suggesting that the HIT selected may be a promising drug candidate for further optimization studies. [6] Other selected ligands subjected to molecular docking gave values ranging from -7.4 to -8.18 Kcal/mol. Ketoconazole is the most effective antifungal because it inhibits the sterol biosynthesis enzyme. [15] The present docking results are consistent because it is an established drug. None of the investigated Imidazoles has surpassed Ketoconazole; all compounds have comparable values. It is observed that the imidazole scaffold is an important pharmacophore that can form favorable interactions within the receptor-binding pocket. [3] Optimizing molecular functional groups or atoms may further enhance hydrophobic interactions, hydrogen bonding, and π-stacking, ultimately increasing the biological activity of the antifungal agent. [16] The computational toxicity assessment using C. tox software, powered by Neurosnap–an AI–based platform–demonstrated that the score ranged from 0.52 to 0.62, with low synthetic accessibility. [9] The molecules are feasible given chemical synthesis. None of the compounds exhibited high toxicity. The ligand C 19 56676630 combined the docking and toxicity scores and was considered a HIT compound in this study. Descriptive statistical analysis indicated stable protein– ligand binding. Mean binding energy was -749 Kcal/mol with a very low value of variation. [10] Minor structural alteration may prove beneficial. Although Ketoconazole possesses the strongest binding affinity, the ligands under study gave comparable binding affinity, toxicity score, and pharmacophoric parameters. [11] Further investigations to optimize HITS-to-leads will yield highly effective antifungal drug candidates that can be tested in vivo and in clinical trials to establish their potency and safety. In vitro antifungal assays may substantiate Insilco study results. The statistical evaluation of the docking scores for Imidazoles indicates consistency and reliability. [10] Only descriptive statistics may be included.
CONCLUSION
A molecular docking study based on an Artificial Intelligence tool was performed on six imidazole compounds against the antifungal target protein 2VOM. Ketoconazole, a clinically established antifungal drug, was used as a reference standard for this study. The values of imidazole ones were compared with those obtained with Ketoconazole. The values indicated good activity prediction, and the compound CID 56676630 exhibited the maximum negative binding energy, indicating stable binding, and toxicity scores indicating druggability. Hence, this compound was selected as an HIT and can be further optimized through chemical structural modification and tested in vivo and in clinical studies to establish it as a new drug in accordance with drug-discovery protocols. The standard Ketoconazole (-9.43 Kcal/mol) and the HIT CID 56676639 (-8.18 kcal/mol) are effective as per in silico testing, e-toxicity, and ADME analysis. Hence, it can be concluded that the imidazole–one scaffold is quite comparable to the nitroimidazole ring scaffold as an antifungal agent, as per in Silico predictions. It can be affirmed that imidazoles possess pi-pi interactions, hydrogen-bonding, and hydrophobic sites that may contribute to stable and effective binding to fungal target receptors. [17] Hence, further optimization of chemical structure or discovery of new chemical entities can be pursued to establish novel antifungal agents with good ADME parameters, lower toxicity, and an acceptable drug profile for future drug development, supported by in vivo and clinical studies. [18-24]
Conflict of Interest:
The authors declare that they have no affiliations or involvement with any organization or entity with a financial interest in the subject matter or materials discussed in this manuscript.
Acknowledgement
We acknowledge the facility provided by the Medical College, Kottayam, for this study.
REFERENCES

Thomas Kurian*, In Silico Molecular Docking And Dynamic Simulation Analysis Of Imidazole-5-Ones And 4- Ones For Antifungal Activity Against Ketoconazole-Bound Fungal Target, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 7, 1899-1908. https://doi.org/ 10.5281/zenodo.21277582
 10.5281/zenodo.21277582
10.5281/zenodo.21277582