
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
HSBPVT'S GOI Faculty of Pharmacy, Kashti, Maharashtra, India.
Histamine H3 receptor antagonists have emerged as promising therapeutic agents for the treatment of central nervous system disorders. In this study, a series of non-imidazole histamine H3 receptor antagonists previously identified through quantitative structure–activity relationship (QSAR) modeling were selected for further computational evaluation. The selected compounds were subjected to comprehensive in silico ADMET analysis to assess their pharmacokinetic and toxicity profiles. Key parameters, including absorption, blood–brain barrier permeability, metabolic stability, cytochrome P450 interactions, and toxicity risks, were systematically evaluated. The results revealed that several compounds exhibited favorable pharmacokinetic properties, including good oral absorption and high predicted brain penetration. Moreover, most compounds showed low toxicity potential and minimal risk of significant drug–drug interactions. These findings demonstrate that QSAR-derived non-imidazole histamine H3 receptor antagonists possess suitable drug-like characteristics for further development. Overall, this study highlights the importance of in silico ADMET profiling as an efficient approach for prioritizing lead compounds and guiding future optimization in drug discovery.
Histamine is a biologically active amine that plays a significant role in various physiological processes, including neurotransmission, immune response, and regulation of the sleep–wake cycle [1]. It exerts its effects through four distinct G-protein-coupled receptor subtypes, namely H1, H2, H3, and H4 receptors [2]. Among these, the histamine H3 receptor is predominantly expressed in the central nervous system and functions as a presynaptic autoreceptor that modulates the release of histamine and other neurotransmitters such as dopamine, serotonin, and acetylcholine [3]. Due to its regulatory role in neurotransmission, the H3 receptor has emerged as an important therapeutic target for neurological disorders, including narcolepsy, Alzheimer’s disease, and attention deficit hyperactivity disorder [4,19,20].
Early development of H3 receptor antagonists primarily focused on imidazole-based compounds; however, these molecules exhibited several limitations, including poor blood–brain barrier penetration, rapid metabolism, and inhibition of cytochrome P450 enzymes [5]. These drawbacks restricted their clinical applicability and prompted the development of non-imidazole derivatives as alternative therapeutic candidates [6]. Non-imidazole H3 receptor antagonists, particularly those containing piperidine and related scaffolds, have demonstrated improved pharmacokinetic properties and enhanced central nervous system activity [7]. Non-imidazole H3 receptor antagonists have been developed to overcome limitations such as cytochrome P450 inhibition associated with imidazole derivatives [21].
Advances in computational chemistry have significantly accelerated drug discovery processes by enabling the prediction of biological activity and pharmacokinetic behavior of chemical compounds. Quantitative structure–activity relationship (QSAR) modeling is a widely used approach that establishes mathematical correlations between molecular descriptors and biological activity, thereby facilitating the identification of key structural features responsible for activity [8]. In a previously reported study, QSAR modeling combined with molecular docking analysis was successfully employed to identify structural determinants influencing the activity of non-imidazole H3 receptor antagonists [9].
Despite the availability of QSAR-derived compounds with promising biological activity, their pharmacokinetic and toxicity profiles remain critical factors for further development. In this context, in silico ADMET analysis has become an essential tool for early-stage drug evaluation [10]. These computational methods allow rapid screening of compounds for drug-likeness, bioavailability, metabolic stability, and safety, thereby reducing the risk of late-stage failure in drug development. In the present study, non-imidazole histamine H3 receptor antagonists previously identified through QSAR modelling were selected for further evaluation using in silico ADMET analysis. The aim of this work is to assess their pharmacokinetic properties and toxicity profiles in order to identify potential lead candidates with favorable drug-like characteristics. Early ADMET screening has been shown to significantly reduce late-stage drug failure rates [22]. Optimizing BBB permeability is essential to ensure effective CNS drug delivery while minimizing adverse effects [23]. In silico ADMET evaluation plays a crucial role in early drug discovery by predicting pharmacokinetic and toxicity profiles and reducing the risk of late-stage failure [15]. CNS activity was evaluated based on blood–brain barrier permeability, an important factor influencing drug action in the brain [17]. Drug-likeness was assessed using Lipinski’s rule of five [14]. The best candidate was identified based on favorable pharmacokinetic properties, low toxicity risk, and compliance with drug-likeness criteria, as commonly applied in rational drug design [18] .
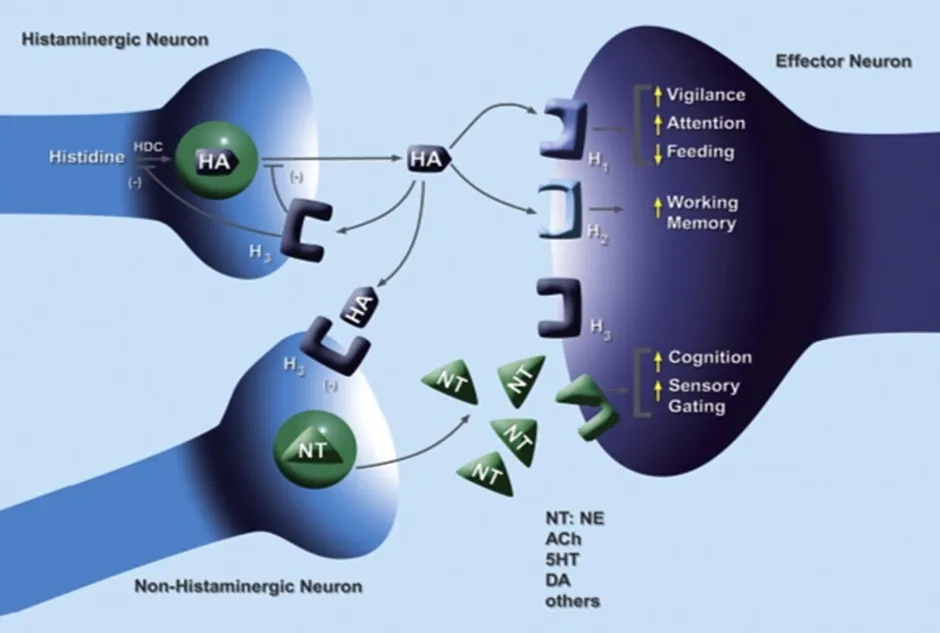
FIG 1: Role of histamine H3 receptors in regulating neurotransmitter release and central nervous system functions. [25]
MATERIAL AND METHODS:
1. Selection of compound:
A total of 56 non-imidazole histamine H3 receptor antagonists were selected from a previously reported QSAR and molecular docking study[9].The were chosen based on their reported biological activity. Structural diversity,and availability of well-defined chemical structures.The dataset was considered suitable for further in silico analysis due to its validated QSAR-derived activity and relevance to central nervous system drug development.
The selected molecules possess non-imidazole scaffolds, which are known to overcome the limitations associated with imidazole-based compounds, such as poor blood–brain barrier penetration and undesirable cytochrome P450 inhibition [5,6]. The dataset includes compounds with well-defined chemical structures and validated activity data, making them suitable for in silico evaluation. Furthermore, the selection was guided by their relevance to central nervous system drug discovery, particularly for evaluating cardiotoxicity and central nervous system activity through ADMET prediction. This dataset was therefore considered appropriate for identifying potential lead candidates with favorable pharmacokinetic and safety profiles.
Table 1: Molecular structures and corresponding IUPAC names and SMILES of selected compounds
|
Sr. No. |
Structure |
IUPAC Name |
Smiles |
|
1 |
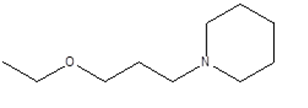
|
1-(3-ethoxypropyl)piperidine
|
CCOCCCN1CCCCC1
|
|
2 |

|
1-(3-propoxypropyl)piperidine
|
CCCOCCCN1CCCCC1
|
|
3 |
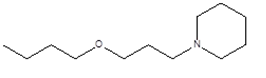
|
1-(3-butoxypropyl)piperidine
|
CCCCOCCCN1CCCCC1
|
|
4 |
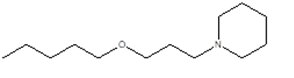
|
1-(3-pentoxypropyl)piperidine
|
CCCCCOCCCN1 CCCCC1
|
|
5 |
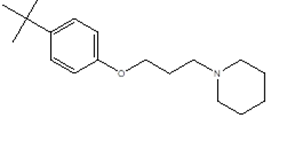
|
1-[3-(4-tert-butylphenoxy)propyl]piperidine
|
CC(C)(C)C(C=C2)= CC=C2OCCCN1CCCCC1
|
|
6 |
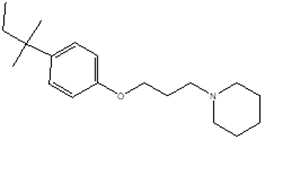
|
1-[3-[4-(2-methylbutan-2-yl)phenoxy]propyl]piperidine
|
CC(CC)(C)C(C=C2)= CC=C2OCCCN1CCCCC1
|
|
7 |

|
1-[[4-[(4-methylphenoxy)methyl]phenyl]methyl]piperidine
|
CC1=CC=C(OCC2=CC=C (CN3CCCCC3)C=C2)C=C1 |
|
8 |

|
1-[[4-[(4-chlorophenoxy)methyl]phenyl]methyl]piperidine
|
ClC1=CC=C(OCC2=CC=C (CN3CCCCC3)C=C2)C=C1
|
|
9 |

|
1-[[4-[(4-methylphenyl)methoxy]phenyl]methyl]piperidine
|
CC1=CC=C(COC2=CC=C (CN3CCCCC3)C=C2)C=C1 |
|
10 |
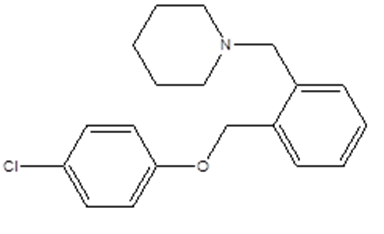
|
1-[[2-[(4-chlorophenoxy)methyl]phenyl]methyl]piperidine
|
ClC1=CC=C(OCC2=CC=CC= C2CN3CCCCC3)C=C1 |
|
11 |

|
1-[3-(3-phenylpropoxy)propyl]-4-propylpiperidine
|
CCCC(CC2)CCN2CCCO CCCC1=CC=CC=C1 |
|
12 |

|
4-butyl-1-[3-(3-phenylpropoxy)propyl]piperidine
|
CCCCC(CC2)CCN2CCCO CCCC1=CC=CC=C1
|
|
13 |

|
4-benzyl-1-[3-(3-phenylpropoxy)propyl]piperidine
|
C1(CCCOCCCN2CCC (CC3=CC=CC=C3)CC2) =CC=CC=C1
|
|
14 |
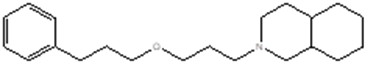
|
2-[3-(3-phenylpropoxy)propyl]-3,4,4a,5,6,7,8,8a-octahydro-1H-isoquinoline |
C1(CCCOCCCN2CC (CCCC3)C3CC2)=CC=CC=C1 |
|
15 |

|
1-[2-(3-chlorophenoxy)ethyl]-4-piperidin-1-ylpiperidine
|
ClC1=CC=CC(OCCN2CCC (N3CCCCC3)CC2)=C1 |
|
16 |

|
1-[2-(3-methoxyphenoxy)ethyl]-4-piperidin-1-ylpiperidine
|
COC1=CC=CC(OCCN2CCC (N3CCCCC3)CC2)=C1
|
|
17 |
|
4-piperidin-1-yl-1-[2-[3-(trifluoromethyl)phenoxy]ethyl]piperidine
|
FC(F)(F)C1=CC(OCCN2CCC (N3CCCCC3)CC2)=CC=C1 |
|
18 |

|
1-[2-(4-methylphenoxy)ethyl]-4-piperidin-1-ylpiperidine
|
CC1=CC=C(OCCN2CCC (N3CCCCC3)CC2)C=C1 |
|
19 |

|
1-[2-(4-chlorophenoxy)ethyl]-4-piperidin-1-ylpiperidine
|
ClC1=CC=C(OCCN2CCC (N3CCCCC3)CC2)C=C1 |
|
20 |

|
1-[3-(4-chlorophenoxy)propyl]-4-piperidin-1-ylpiperidine
|
ClC1=CC=C(OCCCN2CCC (N3CCCCC3)CC2)C=C1 |
|
21 |

|
1-[3-(4-tert-butylphenoxy)propyl]-4-piperidin-1-ylpiperidine
|
CC(C)(C)C1=CC=C (OCCCN2CCC(N3CCCCC3) CC2)C=C1 |
|
22 |

|
1-[3-(3-phenylpropoxy)propyl]piperidine
|
C1(CCCOCCCN2CCCCC2) =CC=CC=C1
|
|
23 |

|
2-(3-piperidin-1-ylpropyl)isoindole-1-carbonitrile
|
N#CC1=C3C(C=CC=C3) =CN1CCCN2CCCCC2
|
|
24 |
|
2-(5-piperidin-1-ylpentyl)isoindole-1-carbonitrile
|
N#CC2=C1C=CC=CC1 =CN2CCCCCN3CCCCC3
|
|
25 |

|
2-(6-piperidin-1-ylhexyl)isoindole-1-carbonitrile
|
N#CC2=C1C=CC=CC1= CN2CCCCCCN3CCCCC3
|
|
26 |
|
4-nitro-N-(4-piperidin-1-ylbutyl)-2,1,3-benzoxadiazol-7-amine
|
O=[N+](C1=CC=C (NCCCCN3CCCCC3)C2= NON=C12)[O-]
|
|
27 |
|
4-nitro-N-(5-piperidin-1-ylpentyl)-2,1,3-benzoxadiazol-7-amine
|
O=[N+](C1=CC=C (NCCCCCN3CCCCC3) C2=NON=C12)[O-]
|
|
28 |
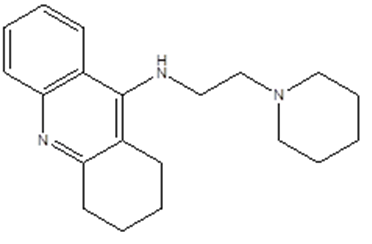
|
N-(2-piperidin-1-ylethyl)-1,2,3,4-tetrahydroacridin-9-amine
|
C13=C(C=CC=C3)C (NCCN2CCCCC2)=C4C(CCCC4)=N1
|
2. In Silico ADMET Prediction:
Pharmacokinetic and toxicity properties were predicted using SwissADME and Toxicity assessment was performed using ProTox-II. The SMILES notation of each compound was used as input.The analysis focused on key parameters, including absorption, distribution, metabolism, excretion, and toxicity. SwissADME was used to evaluate parameters such as gastrointestinal absorption, BBB permability, and drug linkeness.[11] Toxicity profiles, including hepatotoxicity, mutagenicity, and acute toxicity (LD50), were predicted using ProTox-II.[25] Sedative potential was assessed indirectly through blood–brain barrier permeability, as compounds with higher permeability are more likely to exhibit central nervous system activity.Drug-likeness was evaluated using Lipinski’s rule of five which considers molecular weight, lipophilicity (LogP), hydrogen bond donors, and hydrogen bond acceptors. Compounds satisfying these criteria were considered to have favorable oral bioavailability and were prioritized for further analysis. [14] Each identified compound was first assessed and then evaluated against Lipinski’s Rule of Five for drug-likeness. This rule proposed by Christopher A. Lipinski in 1997, is used to assess a chemical compound's pharmacological and ADME properties to determine its potential as an orally active drug.
Toxicity Classes in ProTox-II:
The results obtained from swissADME and ProTox-II were compiled and compared to evaluate the pharmacokinetic behavior, central nervous system activity, and safety profile of the compounds. ADMET evaluation was performed using standard parameters, where molecular weight (<500 Da), LogP (1–5), hydrogen bond donors (≤5), and acceptors (≤10) followed Lipinski’s rule. TPSA (<140 Ų) and high gastrointestinal absorption indicated good permeability. Blood–brain barrier permeability was required for CNS activity, Higher LD₅₀ values and toxicity classes 4–6 indicated safer compounds, whereas low hERG inhibition, inactive Ames toxicity, and low hepatotoxicity were considered essential for safety.
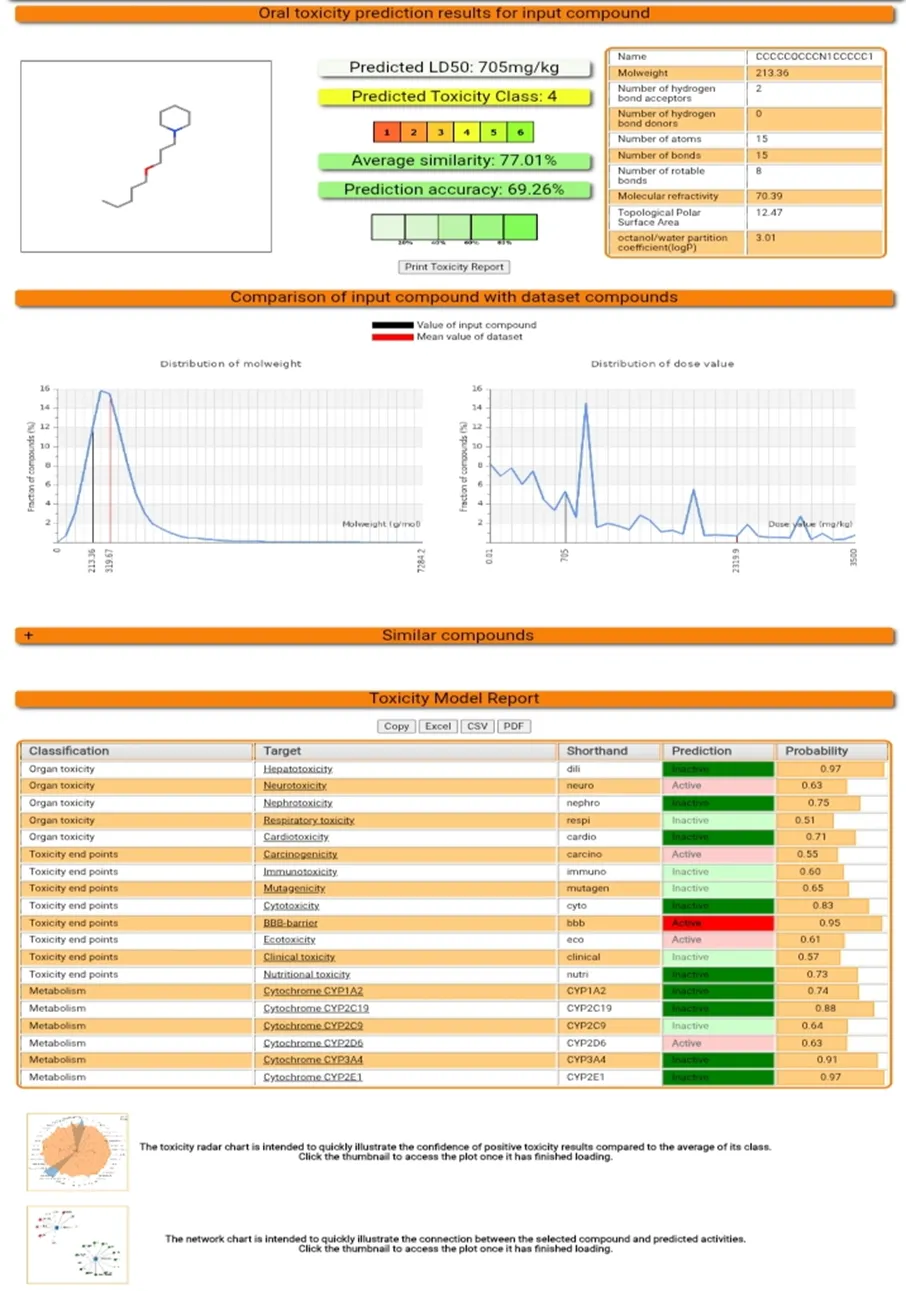
Fig 2: Molecule 4 Toxicity Prediction on Protox -II.
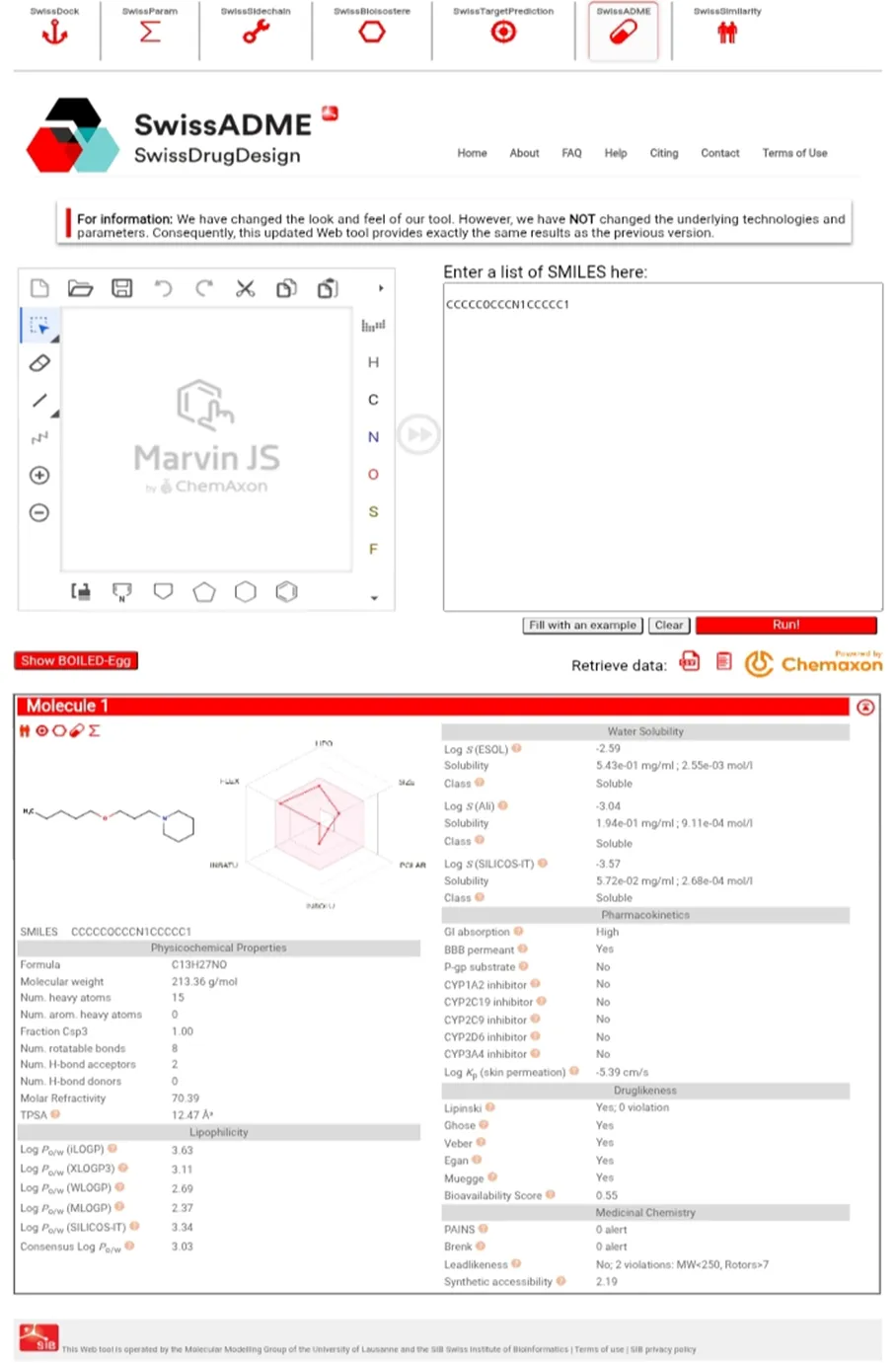
Fig 3: Molecule 4 ADME Prediction on swissADME.
3. Data Analysis:
The predicted ADMET parameters results were compiled and comparatively analyzed. Compounds demonstrating low cardiotoxicity, acceptable blood–brain barrier permeability, favorable pharmacokinetic properties, and minimal toxicity were considered as potential lead candidates. The best compound was identified based on overall performance across all evaluated parameters. Data Analysis The predicted ADMET parameters obtained from SwissADME and ProTox-II were compiled and systematically analyzed. All compounds were compared based on their pharmacokinetic and toxicity profiles. Special emphasis was placed on cardiotoxicity and central nervous system activity.
ADMET RESULT:
Table 2: Predicted ADMET parameters and toxicity profile of selected non-imidazole histamine H3 receptor antagonists
|
Molecule |
GI Absorption |
BBB Permability |
Log P |
TPSA |
No. of Hdon |
No. of H Acc |
Bioavailability Score |
Limpinski |
LD50 mg/ kg |
Toxicity Class |
Mutagenicity (Probability ) |
Hepatotoxicity (Probability) |
|
01 |
High |
Yes |
2.89 |
12.47 |
0 |
2 |
0.55 |
Yes |
780 |
4 |
0.53 |
0.97 |
|
02 |
High |
Yes |
3.31 |
12.47 |
0 |
2 |
0.55 |
Yes |
780 |
4 |
0.67 |
0.97 |
|
03 |
High |
Yes |
3.39 |
12.47 |
0 |
2 |
0.55 |
Yes |
705 |
4 |
0.97 |
0.74 |
|
04 |
High |
Yes |
3.63 |
12.47 |
0 |
2 |
0.55 |
Yes |
705 |
4 |
0.97 |
0.65 |
|
05 |
High |
Yes |
4.01 |
12.47 |
0 |
2 |
0.55 |
Yes |
267 |
3 |
0.82 |
0.97 |
|
06 |
High |
Yes |
4.01 |
12.47 |
0 |
2 |
0.55 |
Yes |
267 |
3 |
0.86 |
0.95 |
|
07 |
High |
Yes |
3.91 |
12.47 |
0 |
2 |
0.55 |
Yes |
221 |
3 |
0.72 |
0.97 |
|
08 |
High |
Yes |
3.99 |
12.47 |
0 |
2 |
0.55 |
Yes |
331 |
4 |
0.69 |
0.93 |
|
09 |
High |
Yes |
3.91 |
12.47 |
0 |
2 |
0.55 |
Yes |
221 |
3 |
0.72 |
0.97 |
|
10 |
High |
Yes |
3.81 |
12.47 |
0 |
2 |
0.55 |
Yes |
100 |
3 |
0.74 |
0.87 |
|
11 |
High |
Yes |
4.32 |
12.47 |
0 |
2 |
0.55 |
Yes |
600 |
4 |
0.74 |
0.98 |
|
12 |
High |
Yes |
4.55 |
12.47 |
0 |
2 |
0.55 |
Yes |
600 |
4 |
0.81 |
0.98 |
|
13 |
High |
Yes |
4.30 |
12.47 |
0 |
2 |
0.55 |
Yes, 1 vivolation: MLOGP>4.15 |
1408 |
4 |
0.75 |
0.98 |
|
14 |
High |
Yes |
4.31 |
12.47 |
0 |
2 |
0.55 |
Yes |
600 |
4 |
0.64 |
0.97 |
|
15 |
High |
Yes |
4.0 |
15.71 |
0 |
3 |
0.55 |
Yes |
1250 |
4 |
0.68 |
0.89 |
|
16 |
High |
Yes |
3.74 |
24.94 |
0 |
4 |
0.55 |
Yes |
1300 |
4 |
0.69 |
0.92 |
|
17 |
High |
Yes |
3.99 |
15.71 |
0 |
6 |
0.55 |
Yes |
559 |
4 |
0.67 |
0.89 |
|
18 |
High |
Yes |
3.95 |
15.71 |
0 |
3 |
0.55 |
Yes |
559 |
4 |
0.71 |
0.93 |
|
19 |
High |
Yes |
3.92 |
15.71 |
0 |
3 |
0.55 |
Yes |
300 |
3 |
0.68 |
0.89 |
|
20 |
High |
Yes |
4.15 |
15.71 |
0 |
3 |
0.55 |
Yes |
212 |
3 |
0.66 |
0.88 |
|
21 |
High |
Yes |
4.59 |
15.71 |
0 |
3 |
0.55 |
Yes |
267 |
3 |
0.74 |
0.92 |
|
22 |
High |
Yes |
3.73 |
12.47 |
0 |
2 |
0.55 |
Yes |
600 |
4 |
0.73 |
0.98 |
|
23 |
High |
Yes |
3.24 |
31.96 |
0 |
2 |
0.55 |
Yes |
2000 |
4 |
0.52 |
0.89 |
|
24 |
High |
Yes |
3.72 |
31.96 |
0 |
2 |
0.55 |
Yes |
2000 |
4 |
0.50 |
0.93 |
|
25 |
High |
Yes |
4.00 |
31.96 |
0 |
2 |
0.55 |
Yes |
2000 |
4 |
0.50 |
0.93 |
|
26 |
High |
Yes |
3.06 |
100.01 |
1 |
6 |
0.55 |
Yes |
1263 |
4 |
0.92 active |
0.69 |
|
27 |
High |
Yes |
2.89 |
100.01 |
1 |
6 |
0.55 |
Yes |
100 |
3 |
0.92 active |
0.69 |
|
28 |
High |
Yes |
3.43 |
28.16 |
1 |
2 |
0.55 |
Yes |
2100 |
5 |
0.73 active |
0.82 |
RESULT:
Molecule 04 [1-(3-pentoxypropyl)piperidine] exhibited the most favorable ADMET and toxicity profile among all evaluated compounds and was identified as the best lead candidate.
DISCUSSION:
A total of 28 QSAR-derived non-imidazole histamine H3 receptor antagonists were evaluated based on their pharmacokinetic and toxicity profiles. The analysis was performed using data obtained from ADMETlab 3.0 and ProTox-II [12,13].
Drug-Likeness and Pharmacokinetic Behavior
All evaluated compounds showed high gastrointestinal absorption and complied with Lipinski’s rule of five, with hydrogen bond donors (≤5) and acceptors (≤10) within acceptable limits. The LogP values ranged between approximately 2.8 and 4.5, indicating suitable lipophilicity for membrane permeability. The TPSA values were below 140 Ų, suggesting good oral bioavailability and permeability.
CNS Activity and Sedative Potential
All compounds exhibited positive blood–brain barrier permeability, indicating their potential for central nervous system activity. This is essential for histamine H3 receptor antagonists targeting neurological disorders such as Alzheimer’s disease and ADHD.
Toxicity Evaluation
The toxicity profiles were assessed using LD₅₀ values and toxicity class predictions. The LD₅₀ values ranged from 100 to 2100 mg/kg, with higher values indicating lower acute toxicity. Most compounds were classified under toxicity class 3–4, indicating moderate safety.
Mutagenicity analysis revealed that some compounds showed high mutagenic potential, while others exhibited comparatively lower values. Hepatotoxicity predictions indicated variability among compounds, with certain molecules showing lower liver toxicity risk.
Identification of the Safest Molecule
Based on combined evaluation of pharmacokinetic and toxicity parameters, Molecule 04 was identified as the safest candidate. It satisfied all major criteria, including:
Although the mutagenicity value was moderate, the overall safety and pharmacokinetic profile of Molecule 04 was superior compared to the remaining compounds.
CONCLUSION
This study evaluated QSAR-derived non-imidazole histamine H3 receptor antagonists using in silico ADMET and toxicity prediction tools. Most compounds showed good drug-likeness, high gastrointestinal absorption, and the ability to cross the blood–brain barrier, indicating their suitability for CNS-related disorders.
Toxicity analysis revealed variability among compounds, particularly in mutagenicity and hepatotoxicity. Among all candidates, Molecule 04 exhibited the most favorable balance of pharmacokinetic and safety parameters, with acceptable toxicity and better overall ADMET profile.
Overall, the findings highlight the usefulness of computational approaches in identifying safer lead compounds, and the selected molecule may be considered for further experimental validation.
REFERENCES



Shinde Snehal, Tikone Mansi, Yadav Madhuri, In Silico Study of QSAR Derived Non-Imidazole Histamine H3 Receptor Antagonist, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 5, 6843-6855. https://doi.org/10.5281/zenodo.20393510
 10.5281/zenodo.20393510
10.5281/zenodo.20393510