
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1MBBS second year, Department of Microbiology, Virology and immunology, Fergana Medical Institute of Public health, Uzbekistan
2Assistant Professor, Department of Microbiology, Virology and immunology, Fergana Medical Institute of Public health Uzbekistan.
Messenger ribonucleic acid (mRNA) vaccine technology has undergone a transformative renaissance driven by the global emergence of SARS-CoV-2 in late 2019. The rapid authorisation of mRNA-based COVID-19 vaccines within twelve months of pathogen identification represents an unprecedented milestone in vaccinology. This review systematically examines the structural architecture, molecular optimisation strategies, immune activation mechanisms, and delivery platforms underpinning mRNA vaccines, with emphasis on COVID-19 applications and emerging clinical horizons. A comprehensive synthesis of peer-reviewed literature was conducted spanning preclinical studies, phase I–III clinical trials, and post-authorisation data across non-replicating mRNA, self-amplifying RNA, circular RNA platforms, and lipid nanoparticle delivery systems. mRNA vaccines offer compelling advantages in speed of manufacture, breadth of immune activation, and platform adaptability. Advances in nucleoside modification, codon optimisation, LNP formulation, and thermal stabilisation address historical limitations. Expanding applications in oncology, HIV, influenza, RSV, and personalised medicine signal a transformative era for nucleic acid-based therapeutics.
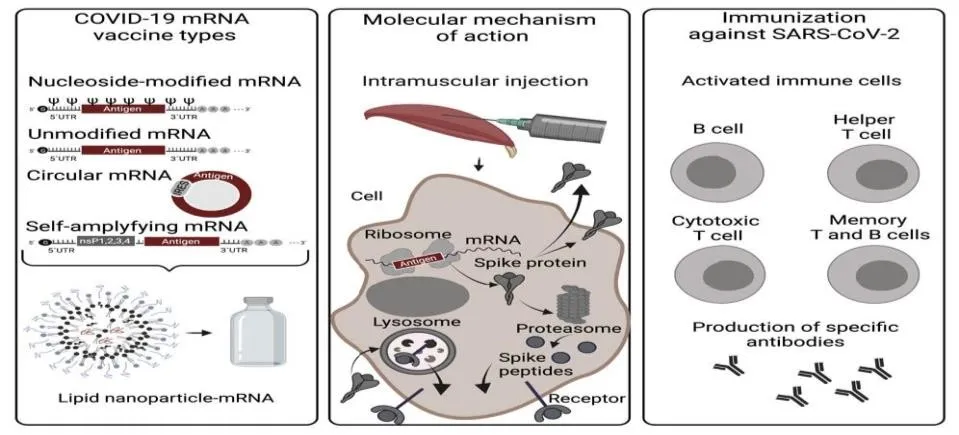
Figure 1. Overview of COVID-19 mRNA vaccine types, intracellular mechanism of action, and adaptive immune cell activation following vaccination. Created with BioRender.com
Vaccination constitutes one of the most cost-effective public health interventions in modern medicine. The emergence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in Wuhan, China in late December 2019, and its designation as a pandemic by the World Health Organization (WHO) in March 2020, created an unprecedented imperative for accelerated vaccine development. Within weeks of the viral genome being deposited publicly in January 2020, multiple candidate vaccines had entered preclinical evaluation—a pace unmatched in vaccine history [1,2]. SARS-CoV-2 is a betacoronavirus harbouring a positive-sense single-stranded RNA genome of ~29,900 nucleotides encoding four principal structural proteins: spike (S), envelope (E), membrane (M), and nucleocapsid (N). The spike glycoprotein, which mediates receptor-binding to human ACE2, became the primary immunogenic target across most vaccine platforms [3,4].

Figure 2. Schematic of the SARS-CoV-2 full-length RNA genome (~29.9 kb) showing ORF1a/b encoding non-structural proteins, four structural genes (S, E, M, N), and the S protein domain map from N-terminal domain (NTD) through the cytoplasmic tail (CT). The receptor-binding domain (RBD) mediates ACE2 engagement.
Historically, vaccine development from discovery to licensure spans ten to fifteen years. The COVID-19 pandemic compelled a paradigm shift, enabling emergency use authorisation (EUA) of mRNA vaccines within twelve months of pathogen characterisation. Two nucleoside-modified mRNA vaccines— BNT162b2 (Pfizer-BioNTech) and mRNA-1273 (Moderna/NIAID)—demonstrated greater than 90% protective efficacy in phase III clinical trials, representing a watershed moment for mRNA technology [5,6]. This review provides a comprehensive synthesis of the structural biology, molecular engineering principles, immunological mechanisms, delivery system innovations, and translational applications of mRNA vaccine platforms. We also examine persistent challenges and future research directions extending mRNA technology into oncology, autoimmune disorders, and personalised medicine.
Table 1. Key historical milestones in vaccine development leading to the mRNA vaccine era.
|
Year |
Milestone |
Significance |
|
1796 |
Jenner's smallpox vaccination |
Established the foundational principle of protective immunity via inoculation |
|
1885 |
Pasteur's rabies vaccine |
First human use of a live-attenuated viral vaccine |
|
1955 |
Salk inactivated polio vaccine (IPV) |
Demonstrated efficacy of killed-virus vaccines at mass scale |
|
1986 |
Recombinant hepatitis B vaccine |
First licensed recombinant protein subunit vaccine |
|
1989 |
Direct mRNA injection in vivo (Wolff et al.) |
Proved mRNA could be expressed as protein in living muscle tissue |
|
2005 |
Nucleoside-modified mRNA (Kariko & Weissman) |
Showed pseudouridine substitution reduces immunogenicity; Nobel Prize 2023 |
|
2012 |
LNP-mRNA antigen expression in vivo |
First demonstration of efficient LNP-delivered mRNA antigen expression |
|
2020 |
EUA for BNT162b2 and mRNA-1273 |
First licensed mRNA vaccines for human use; >90% efficacy vs COVID-19 |
Molecular Architecture of mRNA Vaccines
An mRNA vaccine molecule is a synthetically manufactured single-stranded RNA construct designed to mimic endogenous mature eukaryotic mRNA. Functional mRNA vaccines are synthesised by in vitro transcription (IVT), employing phage-derived RNA polymerases acting upon linearised double-stranded DNA templates. The resulting transcript incorporates five fundamental structural elements, each playing an indispensable role in determining vaccine immunogenicity, translational efficiency, and in vivo stability [4,13].
Table 2. Structural components of mRNA vaccines and their respective biological functions.
|
Structural Element |
Location |
Function |
|
5′ Cap (Cap 1 / m7G) |
5′ terminus |
Prevents exonuclease degradation; recruits eIF4E for ribosomal assembly; reduces innate immune sensing |
|
5′ UTR |
Upstream of ORF |
Guides ribosomal scanning; Kozak sequence optimises AUG start codon recognition |
|
Open Reading Frame (ORF) |
Central coding region |
Encodes target antigen; codon optimisation maximises translational efficiency and protein quality |
|
3′ UTR |
Downstream of ORF |
Regulates mRNA stability and decay rate; alpha/betaglobin-derived sequences widely employed |
|
Poly(A) Tail |
3′ terminus |
Inhibits exonuclease activity; recruits PABPs for closedloop translation; optimal ~100–120 adenosines |
|
Modified Nucleosides |
Within ORF & UTRs |
m1Ψ, Ψ, m5C substitutions suppress TLR3/7/8 activation and enhance translational yield |
The 5′ Cap Structure
The 5′ cap is a chemically modified guanosine nucleotide appended to the transcriptional start site through an unusual 5′-to-5′ triphosphate linkage. The minimal cap structure—7-methylguanosine (m7G), designated Cap 0—confers resistance to 5′-exonuclease cleavage and recruits eIF4E to initiate ribosomal assembly. Sequential 2′-O-methylation of the first transcribed nucleotide produces Cap 1, the preferred configuration for contemporary mRNA vaccines, as it substantially reduces IFIT1-mediated innate immune sensing while augmenting translational output [4,7].
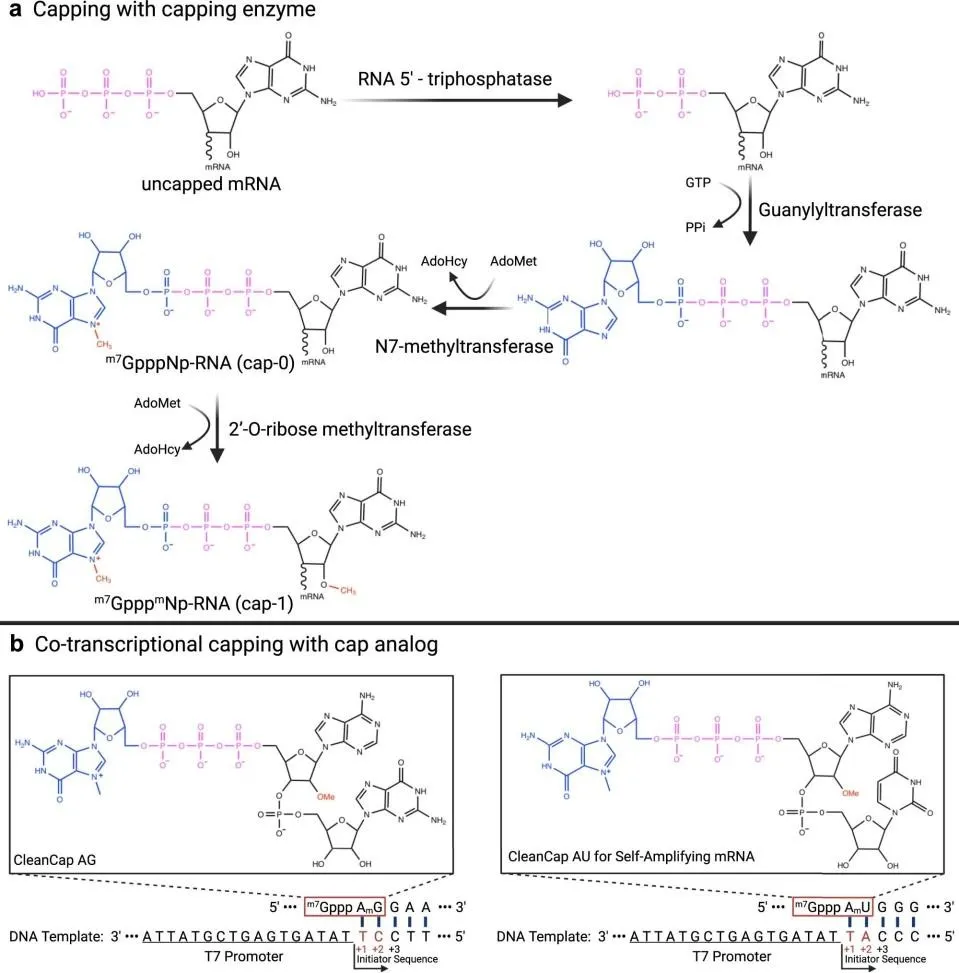
Figure 3. Enzymatic pathways for mRNA 5′ capping. (a) Post-transcriptional three-enzyme reaction generating Cap 0
(m7GpppNp-RNA) then Cap 1 via 2′-O-ribose methyltransferase. (b) Co-transcriptional capping using CleanCap AG and CleanCap AU trinucleotide reagents, yielding Cap 1 at >95% efficiency. Two strategies are employed for in vitro mRNA capping: co-transcriptional incorporation of cap analogue dinucleotides, and post-transcriptional enzymatic capping. Anti-reverse cap analogue (ARCA) prevents reverse incorporation during IVT, yielding homogeneously oriented Cap 0. The CleanCap technology, introduced in 2018, employs a trinucleotide initiating complex to generate a Cap 1 structure cotranscriptionally at efficiencies of 95–99%, and is currently employed in both BNT162b2 and mRNA-1273 [7,13].
Untranslated Regions (UTRs)
The 5′ and 3′ untranslated regions flanking the open reading frame serve as critical regulatory nodes governing translation initiation, mRNA stability, and cellular localisation. The 5′ UTR, typically spanning 53–218 nucleotides, facilitates ribosomal scanning by the pre-initiation complex and contains the Kozak consensus sequence immediately upstream of the AUG start codon. Sequences derived from alpha- and beta-globin mRNAs are commonly incorporated due to their proven capacity to sustain robust protein expression in vivo [4,14]. The 3′ UTR serves as the primary determinant of mRNA half-life in the cytoplasm. AU-rich elements and GU-rich sequences promote mRNA decay and are actively excluded from vaccine designs. Incorporation of stability-enhancing sequences and elimination of destabilising motifs substantially prolongs cytoplasmic mRNA persistence. Precise UTR sequences are considered proprietary intellectual property by vaccine manufacturers, with BioNTech and Moderna using distinct 3′ UTR configurations derived from alpha- and beta-globin regulatory elements [4,13].
Poly(A) Tail
The poly(A) tail—a homopolymeric stretch of adenosine residues at the 3′ terminus—protects the transcript from 3′-to-5′ exonuclease degradation and synergises with the 5′ cap through the poly(A)-binding protein (PABP)–eIF4G interaction to form a translation-competent closed-loop ribonucleoprotein. Empirical data indicate that poly(A) tail length correlates with protein expression up to approximately 120 nucleotides, beyond which marginal increments offer diminishing returns [14]. mRNA-1273 (Moderna) employs a homogeneous poly(A) tail of 100 adenosines. BioNTech adopts a segmented poly(A) architecture—A30-linker-A70—in which two poly(A) sequences are joined by a tennucleotide UGC linker, reportedly extending mRNA half-life and enhancing translational efficiency relative to long-chain continuous poly(A) sequences [4,13].
Classification of COVID-19 mRNA Vaccine Platforms
mRNA-based vaccines are categorised into three principal classes: non-replicating mRNA (nr-mRNA), self-amplifying mRNA (saRNA), and circular RNA (circRNA). Each offers a distinct balance of dose requirement, antigen expression duration, immunogenicity profile, and manufacturing complexity [4,21].
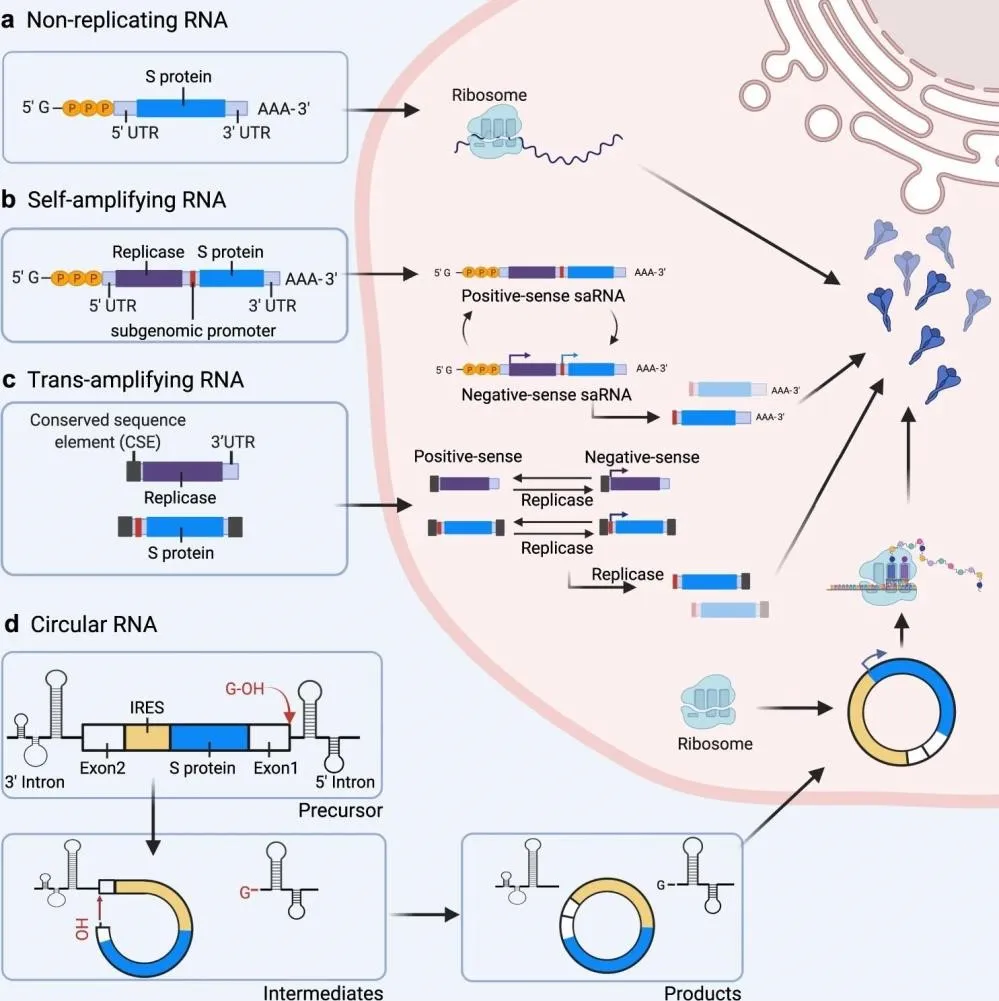
Figure 4. Classification and intracellular behaviour of the four principal mRNA vaccine platforms: (a) non-replicating mRNA directly translated by host ribosomes; (b) self-amplifying RNA (saRNA) encoding a viral replicase enabling cytoplasmic RNA amplification; (c) trans-amplifying RNA (taRNA) separating replicase and antigen into two molecules; (d) circular RNA (circRNA) utilising IRES-driven cap-independent translation with exonuclease resistance.
Non-Replicating mRNA (nr-mRNA) Vaccines
Non-replicating mRNA vaccines represent the most clinically advanced platform and comprise all currently marketed COVID-19 mRNA products. These constructs encode solely the target antigen— typically the prefusion-stabilised full-length SARS-CoV-2 spike protein—flanked by optimised 5′ and 3′ UTRs, capped with Cap 1, and terminated by a poly(A) tail. Following intramuscular injection and endosomal uptake, the mRNA is released into the cytoplasm where host ribosomes translate it into the encoded antigen. The expressed protein is displayed on MHC class I and II molecules and activates professional antigen-presenting cells in draining lymph nodes [4,7]. The advantages of nr-mRNA vaccines include structural simplicity, ease of large-scale IVT manufacture, rapid sequence modification in response to emerging variants, and absence of genomic integration risk. The principal limitation relates to thermolability necessitating ultra-cold storage, and the transient duration of antigen expression [6,13].
Self-Amplifying mRNA (saRNA) Vaccines
Self-amplifying RNA vaccines are derived from positive-strand RNA virus genomes, most commonly alphaviruses such as Semliki Forest virus (SFV), Venezuelan equine encephalitis virus (VEEV), and Sindbis virus (SIN). The saRNA construct retains viral replicase-encoding non-structural proteins (nsP1– nsP4) that together form an RNA-dependent RNA polymerase (RDRP) capable of amplifying genomic and subgenomic RNA templates intracellularly. Viral structural protein genes are replaced by the vaccine antigen sequence, rendering the construct incapable of producing infectious virions [21,22].
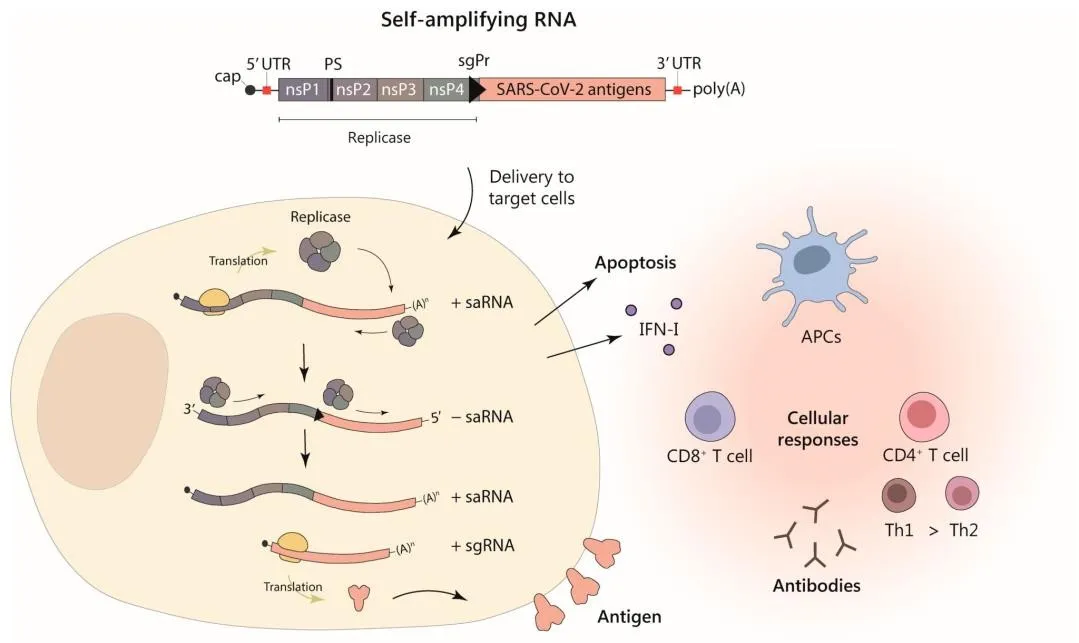
Figure 5. Alphavirus-based self-amplifying RNA (saRNA) vector expressing SARS-CoV-2 antigens. The saRNA encodes a replicase complex (nsP1–4) that amplifies both genomic and subgenomic RNA, enabling high-level antigen expression from lowdose delivery. Cellular immune responses include type I IFN induction, apoptosis, and activation of humoral and CD4+/CD8+ T cell responses via antigen-presenting cells (APCs).
Upon entry into the cytoplasm, saRNA is first translated to produce the RDRP complex, which amplifies both positive-sense genomic RNA and a smaller subgenomic RNA encoding the antigen. This cascade produces considerably higher antigen expression from substantially lower doses—typically 1–10 μg compared to 30–100 μg for nr-mRNA—making saRNA advantageous for mass immunisation. Two saRNA COVID-19 vaccines have received regulatory approval: GEMCOVAC-OM in India and ARCT-154 in Japan [21,22].
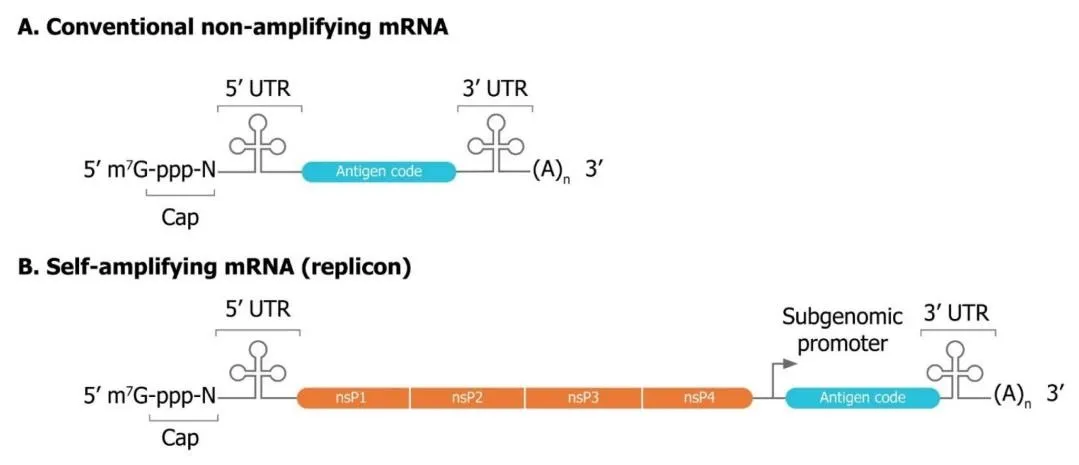
Figure 6. Structural comparison of conventional non-amplifying mRNA (A) and self-amplifying mRNA (B). Both share cap, 5′
UTR, 3′ UTR, and poly(A) tail, but saRNA additionally encodes four non-structural replicase proteins (nsP1–4) and a subgenomic promoter, enabling intracellular RNA amplification at substantially lower administered doses.
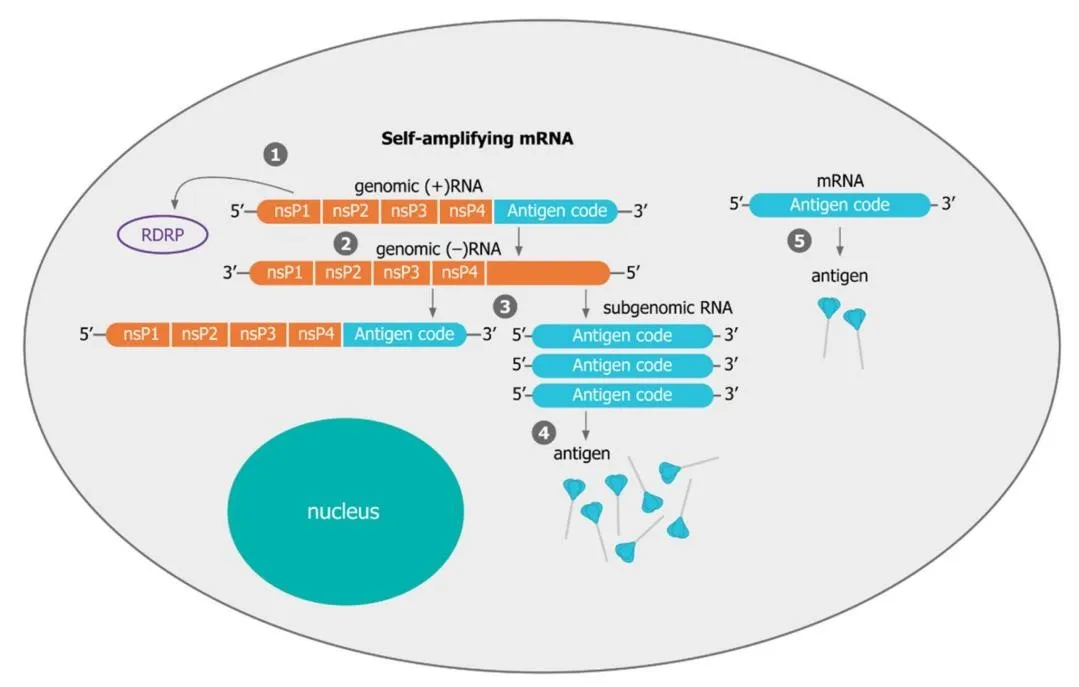
Figure 7. Schematic comparing intracellular mechanisms of saRNA versus conventional mRNA. In saRNA, the translated replicase produces multiple copies of both genomic and subgenomic RNA, generating far more antigen per delivered molecule than non-amplifying mRNA.
Trans-amplifying RNA (taRNA) systems—a further innovation—separate the replicase-encoding and antigen-encoding components into two distinct RNA molecules delivered together. This bipartite architecture allows independent optimisation of each component and reduces the overall antigen-encoding transcript size, facilitating more efficient encapsulation [22].
Circular RNA (circRNA) Vaccines
Circular RNA possesses a covalently closed ring structure lacking free 5′ and 3′ termini, conferring remarkable resistance to exonuclease-mediated degradation and substantially extended half-life compared to linear mRNA. Translation is initiated by internal ribosome entry sites (IRES) rather than conventional 5′ cap-dependent mechanisms. Preclinical studies have demonstrated that LNP-encapsulated circRNA vaccines encoding the RBD of SARS-CoV-2 Omicron maintain robust expression for at least two weeks at ambient temperature (~25°C), highlighting their potential as thermostable candidates [4,14].
Mechanism of Immune Activation by mRNA Vaccines
Antigen Expression and Processing
Following intramuscular administration, mRNA-LNP complexes are taken up by cells at the injection site—predominantly myocytes, fibroblasts, and infiltrating dendritic cells—through endocytic pathways including macropinocytosis and clathrin-mediated endocytosis. Acidification of endosomal compartments triggers ionisable lipid protonation, destabilising the endosomal membrane and enabling cytosolic mRNA release. Once in the cytoplasm, the mRNA is translated into the SARS-CoV-2 prefusion-stabilised spike protein [4,25]. Synthesised antigen follows the secretory pathway, undergoing glycosylation in the endoplasmic reticulum before being transported to the cell surface for MHC class I presentation to CD8+ cytotoxic T lymphocytes (CTLs). Simultaneously, secreted antigen is internalised by APCs and presented on MHC class II molecules to CD4+ helper T lymphocytes. mRNA-LNP vaccines preferentially drain to lymph nodes where they activate germinal centre B cell responses and follicular helper T cell (Tfh) differentiation, driving the affinity maturation necessary for high-quality neutralising antibody production [5,25].
Innate Immune Response
The innate immune response involves recognition of exogenous mRNA by pattern recognition receptors (PRRs). Single-stranded RNA activates endosomal TLR7 and TLR8 in plasmacytoid dendritic cells and macrophages; double-stranded RNA generated during IVT may engage TLR3, RIG-I, and MDA5. These activations trigger type I interferon (IFN-α/β) responses, NF-κB activation, and release of proinflammatory cytokines including TNF-α, IL-6, and IL-1β [4,17].
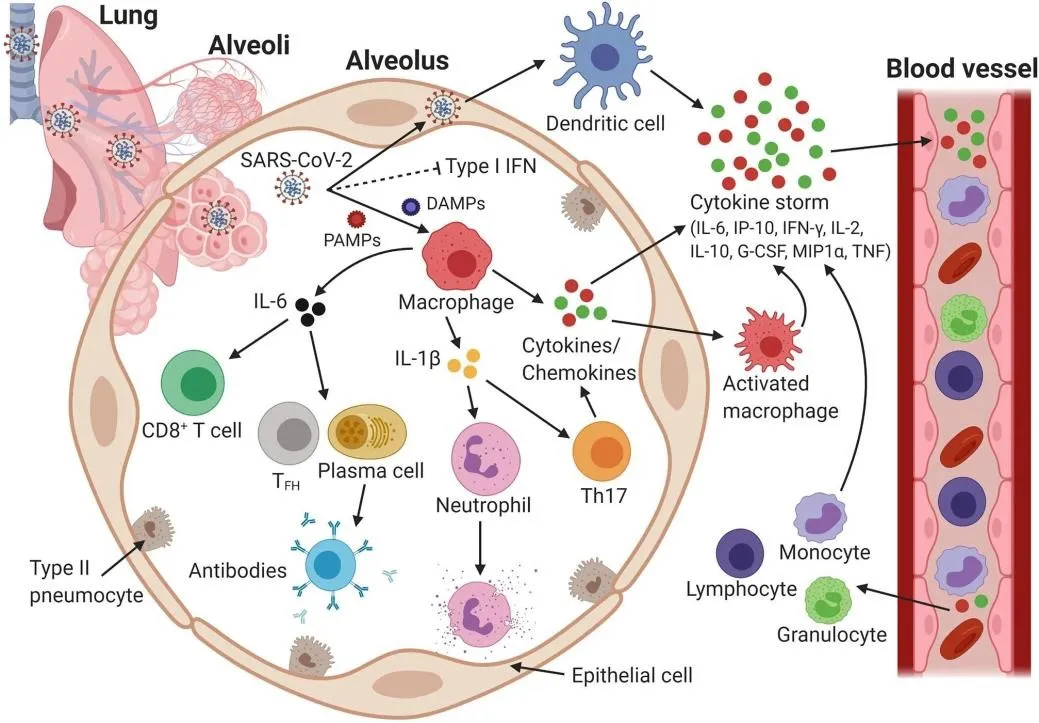
Figure 8. Host immune response and immunopathology during severe SARS-CoV-2 infection. SARS-CoV-2 suppresses type I IFN responses, enabling uncontrolled viral replication. PAMPs and DAMPs activate macrophages and epithelial cells to release cytokines and chemokines, initiating a cytokine storm (IL-6, IP-10, IFN-γ, G-CSF, TNF-α). The resulting feedback loop drives multi-organ damage via CD8+ T cells, Th17 cells, and neutrophils.
Nucleoside modifications—particularly substitution of all uridines with N1-methylpseudouridine (m1Ψ)— substantially dampen innate immune sensing while preserving robust adaptive immune activation. LNPs themselves contribute an intrinsic adjuvant effect, inducing local release of chemokines including CCL2, CCL3, CXCL1, and IL-6 at the injection site [5,26].
Adaptive Immune Response
Adaptive immunity generated by mRNA vaccines encompasses both humoral and cellular arms. On the humoral side, B cells encountering spike antigen within germinal centres undergo extensive somatic hypermutation and affinity selection, producing high-affinity IgG antibodies targeting primarily the receptor-binding domain (RBD). Neutralising antibodies block spike-ACE2 interaction, while nonneutralising antibodies contribute to Fc-mediated effector functions [17]. CD4+ T helper cells provide cognate help to B cells for class-switching and affinity maturation. CD8+ CTLs are capable of recognising and eliminating spike-expressing cells. Long-term immunological memory is established through differentiation of memory B cells, central memory CD4+ T cells, and memory CD8+ T cells, providing durable protection against re-infection [17,27].
Delivery Systems for mRNA Vaccines
The intrinsic physicochemical properties of mRNA—large molecular weight (10⁴–10⁶ Da), anionic charge, susceptibility to RNase degradation, and poor cellular membrane permeability—necessitate encapsulation vehicles for effective in vivo administration. Three principal delivery system classes have been evaluated: lipid nanoparticles (LNPs), polymer-based systems, and viral vectors [4,29].
Lipid Nanoparticle (LNP) Delivery
Lipid nanoparticles represent the delivery platform of choice for all currently approved COVID-19 mRNA vaccines, providing high encapsulation efficiency, endosomal escape capability, low immunogenicity, and scalable manufacturing. LNPs are self-assembled multicomponent structures comprising four lipid species in defined molar ratios [4,28].
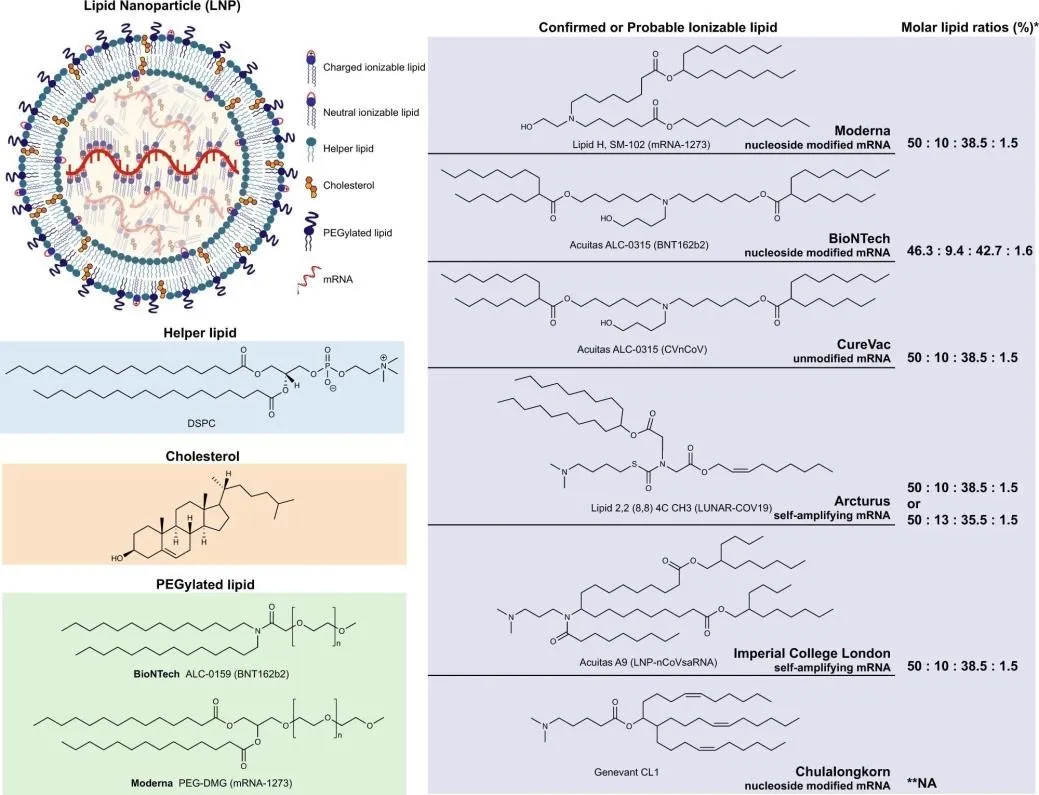
Figure 9. LNP architecture and ionisable lipid chemical structures. LNPs are composed of ionisable lipid (central core), helper lipid and PEGylated lipid (bilayer surface), and cholesterol (distributed throughout). Shown are confirmed ionisable lipid structures for Moderna (SM-102), BioNTech (ALC-0315), CureVac, Arcturus, Imperial College London, and Chulalongkorn University formulations, with molar lipid ratios.
Table 3. Composition, examples, and functional roles of the four LNP lipid components.
|
Component |
Molar Ratio (%) |
Key Examples |
Primary Function |
|
Ionisable Lipid |
30–50% |
ALC-0315 (BNT162b2); SM102 (mRNA-1273); DLinMC3-DMA |
Complexes mRNA at acidic pH; enables endosomal escape upon protonation |
|
Helper Phospholipid |
10–20% |
DSPC; DOPE; ALC-0159 |
Stabilises bilayer; facilitates membrane fusion and cellular uptake |
|
Cholesterol |
30–40% |
Plant-derived or synthetic cholesterol |
Regulates membrane fluidity and permeability; enhances structural integrity |
|
PEGylated Lipid |
1–2% |
PEG2000-DMG (Moderna); ALC-0159 (Pfizer) |
Prevents aggregation; reduces opsonisation; controls particle size |
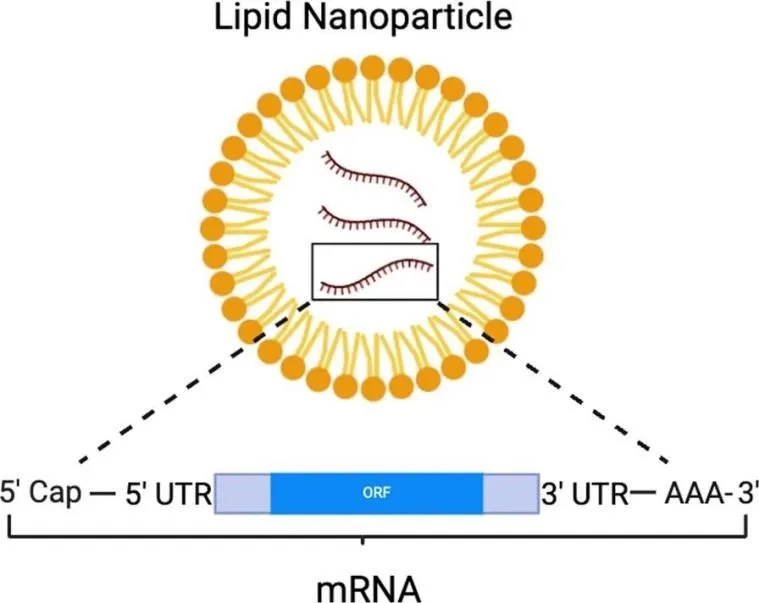
Figure 10. Schematic of mRNA encapsulation within a lipid nanoparticle (LNP). The mRNA molecule (5′ Cap – 5′ UTR – ORF – 3′ UTR – poly(A)) is protected within the LNP bilayer structure, enabling cellular delivery, endosomal escape, and cytoplasmic release. Created with BioRender.com.
The ionisable lipid is the pivotal functional component. At mildly acidic pH (~4.0) during nanoparticle assembly, it bears a positive charge facilitating electrostatic complexation with negatively charged mRNA. At physiological pH (7.4), the lipid returns to neutral, reducing non-specific interactions with serum proteins. Upon endosomal acidification, re-protonation disrupts the endosomal bilayer, enabling mRNA cytoplasmic release [5,7].
BNT162b2 employs ionisable lipid ALC-0315, and mRNA-1273 uses SM-102. Both incorporate biodegradable ester linkages designed to reduce cumulative toxicity upon repeated dosing. PEGylated lipids—present at 1–2% molar ratio—sterically stabilise the nanoparticle surface, preventing aggregation and prolonging circulation half-life [5,28].
Polymer-Based Delivery Systems
Polymeric nanoparticles offer an alternative to lipid-based systems with advantages in structural versatility and surface functionalisation. Cationic polymers including polyethylenimine (PEI), poly(beta-amino esters) (PBAEs), and polyamidoamine (PAMAM) dendrimers condense mRNA through electrostatic interactions and facilitate endosomal escape via the proton sponge mechanism [13,29]. PBAEs have emerged as particularly promising due to biodegradability, reduced cytotoxicity compared to PEI, and efficient mRNA delivery across a range of cell types. Chitosan-based systems have been investigated for intranasal mRNA vaccination, exploiting their ability to transiently open epithelial tight junctions to access nasal-associated lymphoid tissue (NALT). Despite encouraging preclinical results, polymer-based mRNA delivery systems have yet to achieve clinical licensure for vaccines [13,56]. 5.3 Viral Vector Delivery
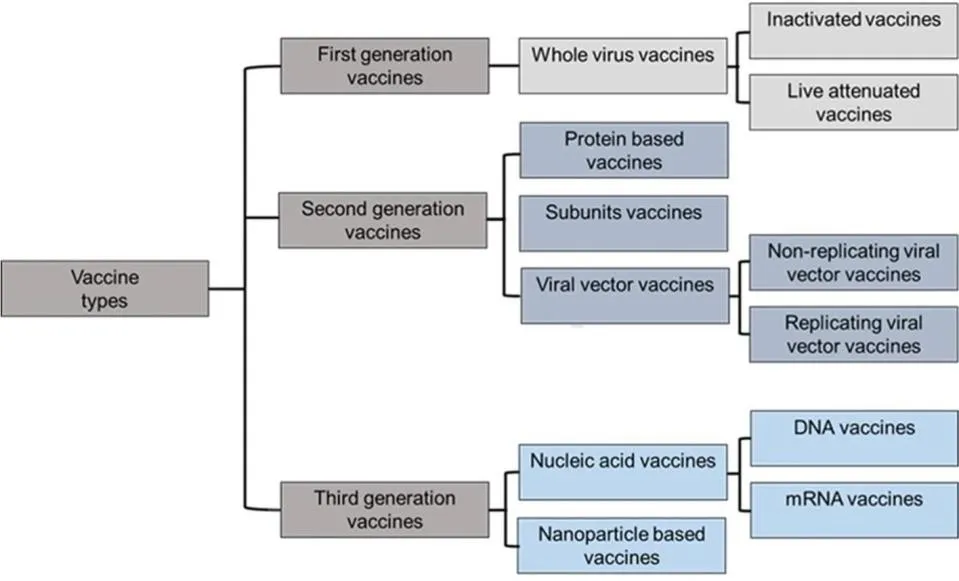
Figure 11. Main vaccine types and platforms for SARS-CoV-2 including whole virus (inactivated and live-attenuated), viral vector (replicating and non-replicating), protein-based (VLP and recombinant subunit), and nucleic acid vaccines (DNA and RNA), highlighting their respective mechanisms of antigen delivery.
Viral vectors leverage natural cellular transduction machinery to deliver antigen-encoding genetic material with high efficiency. Non-replicating viral vector vaccines (VVnr) based on adenoviral scaffolds have achieved widespread clinical use. The adenoviral genome is engineered to delete replication-essential genes (E1/E3 deletions) and insert the SARS-CoV-2 spike-encoding sequence, yielding a vector that infects cells and expresses the antigen but cannot self-replicate [16,31].
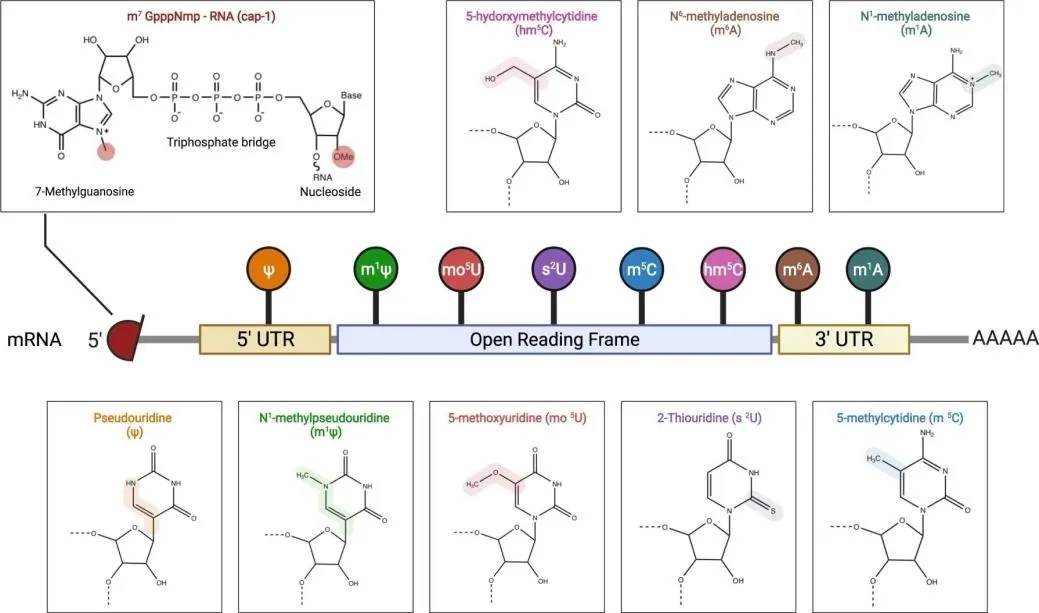
Figure 12. Vaccine front-runner candidates for SARS-CoV-2. Candidates span whole-virus, viral vector, protein-based, and nucleic acid categories, each exploiting different mechanisms to present SARS-CoV-2 antigens to the immune system. Nucleic acid vaccines (DNA and mRNA-LNP) offer the fastest manufacturing adaptability.
Key adenoviral vector COVID-19 vaccines include: ChAdOx1 nCoV-19 (AstraZeneca/Oxford), derived from chimpanzee adenovirus to circumvent pre-existing human immunity; Ad26.COV2.S (Janssen), using human adenovirus type 26; Sputnik V (Gamaleya), employing a heterologous prime-boost strategy with Ad26 and Ad5 vectors; and Ad5-nCoV/Convidecia (CanSino), based on Ad5. Pre-existing vector-specific immunity—particularly widespread anti-Ad5 seroprevalence—remains the principal limitation of adenoviral vector vaccines [6,31].
Molecular Optimisation Strategies
Nucleoside Modifications
The pioneering work of Kariko and Weissman (2005) established that substitution of naturally occurring nucleosides with chemically modified analogues dramatically attenuates innate immune recognition of synthetic mRNA while preserving translational competence. Unmodified exogenous mRNA activates TLR3 (dsRNA), TLR7, and TLR8 (ssRNA), triggering interferon responses that suppress ribosomal activity and accelerate mRNA degradation. Incorporation of modified nucleosides abrogates TLR binding through conformational changes that reduce recognition by the receptor's hydrophobic binding pocket [4,7].
Table 4. Nucleoside modifications employed in mRNA vaccine development and their mechanistic and clinical implications.
|
Modification |
Code |
Mechanism |
Clinical Relevance |
|
N1methylpseudouridine |
m1Ψ |
Suppresses TLR3/7/8 and RIG-I; enhances ribosomal binding affinity |
Used in BNT162b2 & mRNA- 1273; current gold-standard modification |
|
Pseudouridine |
Ψ |
Reduces innate immune signalling; stabilises mRNA secondary structure |
Early Kariko-Weissman formulations; superseded by m1Ψ for most applications |
|
5-methylcytidine |
m5C |
Evades TLR3 and TLR7/8 recognition; reduces dsRNA contaminant sensing |
Used in combination with Ψ in pioneer nucleoside-modified mRNA vaccines |
|
5-methoxyuridine |
mo5U |
Diminishes TLR-mediated sensing; reduces innate immune activation |
Applied in saRNA constructs for reduced inflammatory response |
|
N6-methyladenosine |
m6A |
Modulates mRNA stability and translation efficiency; reduces RNase L activation |
Investigated for improved translational output in therapeutic mRNA |
N1-methylpseudouridine (m1Ψ) has supplanted pseudouridine (Ψ) as the preferred modification in current clinical mRNA vaccines. Studies demonstrated that m1Ψ-modified mRNA administered intradermally produced 20-fold higher protein levels compared to sequence-optimised but unmodified mRNA.
Nucleoside modification also facilitates germinal centre responses and Tfh cell differentiation—processes critical for long-lasting humoral immunity [5,7].
Codon Optimisation
Codon optimisation enhances translational efficiency by adapting the vaccine ORF nucleotide sequence to match host codon usage bias without altering the encoded amino acid sequence. Human cells maintain differential abundances of aminoacyl-tRNAs corresponding to synonymous codons; substituting rare codons with frequently used synonymous alternatives reduces ribosomal pausing and accelerates translational elongation [7,38]. Guanosine-cytidine (GC) enrichment is a widely applied codon optimisation strategy. GC-rich sequences can achieve translational efficiency up to 100-fold greater than GC-poor equivalents. Bioinformatics algorithms such as CDSfold and deep learning-based sequence design tools are increasingly employed to simultaneously optimise translational efficiency, mRNA secondary structure stability, and antigen folding quality [38,39].
Structural Stabilisation
mRNA stability—encompassing chemical integrity and structural conformation—is a paramount determinant of vaccine potency and shelf life. Chemical degradation in aqueous environments proceeds primarily through base-catalysed phosphodiester hydrolysis, nucleobase oxidation, and depurination—all of which generate strand breaks that halt ribosomal translation. Even a single phosphodiester cleavage within a 4,000-nucleotide mRNA molecule can ablate antigen expression [36,41]. Lyophilisation (freeze-drying) removes the aqueous medium responsible for hydrolytic degradation and is the most promising approach for improving mRNA-LNP thermal stability. Current research demonstrates viable storage at 2–8°C for up to 18 months in lyophilised formulations. Secondary structure engineering— designing ORFs to maximise intramolecular base-pairing—reduces enzymatic susceptibility [40,42].
COVID-19 Vaccine Candidates: Platform-Specific Profiles
Table 5. Comparative profiles of major COVID-19 vaccines across different platforms.
|
Vaccine |
Developer |
Platform |
Efficacy (%) |
Storage |
Dosing |
|
BNT162b2 (Comirnaty) |
Pfizer/BioNTech |
mod-mRNA LNP |
~95% |
-60 to -80°C |
2 doses / 21 days |
|
mRNA-1273 (Spikevax) |
Moderna/NIAID |
mod-mRNA LNP |
~94% |
-20 to -25°C |
2 doses / 28 days |
|
CVnCoV (CureVac) |
CureVac |
UnmodmRNA LNP |
~47% |
2°C to 8°C |
2 doses / 28 days |
|
ChAdOx1 (Vaxzevria) |
AstraZeneca/Oxford |
Non-rep. viral vector |
~70% |
2°C to 8°C |
2 doses / 4–12 wks |
|
Sputnik V |
Gamaleya Institute |
Ad26 + Ad5 vector |
~91% |
-18°C |
2 doses (Ad26+Ad5) |
|
CoronaVac |
Sinovac |
Inactivated virus |
50–84% |
2°C to 8°C |
2 doses / 14 days |
BNT162b2 (Pfizer-BioNTech)
BNT162b2 (Comirnaty) is a nucleoside-modified mRNA vaccine encoding the full-length prefusionstabilised SARS-CoV-2 spike protein, formulated in ALC-0315/ALC-0159-containing LNPs. The spike sequence incorporates two proline substitutions at K986 and V987 constraining the protein in the antigenically optimal prefusion conformation. Phase III data across 44,000 participants demonstrated 95.0% (95% CI 90.3–97.6%) efficacy against symptomatic COVID-19 at seven days post-second dose. The vaccine is administered as two intramuscular doses of 30 μg separated by 21 days [23,25].
mRNA-1273 (Moderna)
mRNA-1273 (Spikevax) encodes the same prefusion-stabilised spike protein using an SM-102/PEG2000DMG LNP formulation at 100 μg per injection, administered as two doses separated by 28 days. Phase III COVE trial data across 30,000 participants demonstrated 94.1% (95% CI 89.3–96.8%) vaccine efficacy. The formulation achieves stability at -20°C—a significant advantage over BNT162b2's initial -80°C requirement—with demonstrated stability for up to 30 days at 2–8°C [5,7].
ChAdOx1 nCoV-19 (AstraZeneca)
ChAdOx1 nCoV-19 uses a recombinant replication-deficient chimpanzee adenovirus vector delivering the full-length SARS-CoV-2 spike protein with a tissue plasminogen activator (tPA) signal sequence at the 5′ end. Interim multinational Phase III trials reported pooled efficacy of 70.4% against symptomatic COVID19. Crucially, storage at 2–8°C substantially simplifies cold-chain logistics for global distribution. Postauthorisation surveillance identified rare vaccine-induced immune thrombocytopaenia and thrombosis (VITT/TTS), prompting age-stratified regulatory guidance [23].
CoronaVac (Sinovac)
CoronaVac is an inactivated whole-virus vaccine produced by inoculating SARS-CoV-2 (CZ02 strain) into Vero cells, inactivating with β-propiolactone, and adjuvanting with aluminium hydroxide. It can be stored at 2–8°C for years, conferring major logistical advantages for lower-income settings. Phase III efficacy results ranged from 50.7% (Brazil) to 83.5% (Turkey), reflecting the impact of differing circulating strains, study populations, and definitions of protection [23].
Challenges and Limitations of mRNA Vaccines
Thermal Instability and Cold-Chain Requirements
The thermolability of mRNA-LNP formulations constitutes the most significant logistical barrier to equitable global vaccine distribution. Current approved mRNA vaccines require cold-chain maintenance at -80°C (BNT162b2 at initial rollout) to -20°C (mRNA-1273), imposing enormous infrastructure demands that substantially exceed the capacity of low- and middle-income countries (LMICs). Multiple strategies are being pursued: lyophilisation (demonstrated 18-month shelf life at 2–8°C for mRNA-1647), thermostable excipient matrices, structural RNA engineering to maximise base-pairing, and development of inherently more stable saRNA and circRNA platforms [36,48].
Variant Emergence and Immune Evasion
The emergence of SARS-CoV-2 variants of concern—Alpha (B.1.1.7), Beta (B.1.351), Delta (B.1.617.2), and Omicron (B.1.1.529)—has challenged the durability of vaccine-induced immunity. Omicron, with over 30 spike mutations, demonstrated markedly reduced neutralisation by antibodies elicited by original-strain mRNA vaccines, necessitating rapid development of bivalent booster formulations. The mRNA platform's intrinsic adaptability enabled bivalent boosters targeting ancestral and Omicron BA.4/BA.5 spikes to be authorised within approximately six months of Omicron emergence [50,51].
Regulatory Complexity and Vaccine Equity
The emergency use authorisation pathway, while enabling rapid deployment, raised legitimate questions regarding the completeness of long-term safety data, contributing to hesitancy in some populations. Divergent regulatory requirements between the FDA, EMA, and WHO increase redundancy in approval processes. Vaccine inequity remains a profound global health failure: high-income countries administered multiple booster doses while the majority of low-income country populations remained unvaccinated in 2021–2022. Structural determinants including intellectual property protections, manufacturing concentration, bilateral advance purchase agreements, and cold-chain deficiencies collectively compounded this inequity [48,49].
Clinical Applications of mRNA Technology
The success of mRNA vaccines in COVID-19 has catalysed an extraordinary expansion of mRNA-based therapeutic programmes across oncology, virology, rare disease, and autoimmune medicine. The versatility of the mRNA platform—enabling encoding of virtually any protein of interest within a standardised manufacturing framework—positions it as a potentially transformative modality across multiple indications [24,46].
Table 6. Selected clinical applications of mRNA vaccine technology beyond COVID-19.
|
Disease |
Candidate |
Stage |
Key Findings |
|
COVID-19 |
BNT162b2, mRNA1273 |
Approved (Full + EUA) |
>90% efficacy; robust humoral and cellular immunity |
|
Influenza |
mRNA-1010 (Moderna) |
Phase III |
Quadrivalent; improved HA/NA antigen breadth vs standard vaccines |
|
RSV |
mRNA-1345 (Moderna) |
Approved in some regions |
>80% efficacy against RSV lower respiratory tract disease |
|
HIV |
eOD-GT8 60mer mRNA-LNP |
Phase I (NCT05001373) |
First clinical proof-of-concept for germline-targeting priming |
|
Melanoma |
mRNA-4157/V940 + Pembro |
Phase III (personalised) |
44% reduction in recurrence/death vs pembrolizumab alone |
|
CMV |
mRNA-1647 (Moderna) |
Phase III; lyophilised |
18-month shelf life at 5°C; 69% efficacy in seronegative women |
|
Zika Virus |
mRNA-1893 (Moderna) |
Phase II |
Robust neutralising antibody titres; prM-E protein antigen |
Influenza
Seasonal influenza vaccines require annual reformulation constrained by lengthy egg-based or cell-based manufacturing. mRNA vaccines eliminate pathogen cultivation, enabling sequence-to-vaccine production within weeks and dramatically compressing the window for strain mismatch. Moderna's mRNA-1010 quadrivalent seasonal influenza vaccine candidate demonstrated non-inferior immunogenicity to licensed comparator vaccines in Phase III trials. Combination mRNA vaccines co-expressing influenza and other respiratory pathogen antigens are under active development [54].
Human Immunodeficiency Virus (HIV)
HIV vaccine development has been confounded for four decades by exceptional antigenic variability of the HIV-1 envelope glycoprotein and the difficulty of eliciting broadly neutralising antibodies (bnAbs). The germline-targeting vaccine concept aims to prime bnAb precursor B cells using immunogens designed to bind these rare naive B cell populations with high affinity. The eOD-GT8 60-mer nanoparticle immunogen delivered as nucleoside-modified mRNA-LNP in clinical trial NCT05001373 successfully induced VRC01-class bnAb precursor B cells in recipients—providing the first clinical proof-of-concept for germline-targeting priming [10].
Cancer Immunotherapy
Cancer vaccines leverage mRNA technology to stimulate immune responses against tumour-specific antigens (TSAs) or neoantigens—protein sequences derived from somatic mutations unique to each patient's tumour. This personalised approach requires whole-exome tumour sequencing, computational neoantigen prediction, rapid mRNA synthesis, and patient-specific manufacturing—all achievable within the mRNA platform's flexible framework [10,24]. The most advanced personalised mRNA cancer vaccine programme is mRNA-4157/V940 (Moderna/Merck), encoding up to 34 patient-specific neoantigens identified from resected tumour tissue. In combination with pembrolizumab (anti-PD-1 inhibitor), V940 demonstrated a 44% reduction in recurrence or death versus pembrolizumab alone in Phase II/b KEYNOTE-942 trial in high-risk resected stage II–IV melanoma. BioNTech's autogene cevumeran (BNT122) demonstrated durable polyspecific T cell responses and delayed tumour recurrence over three years in resected pancreatic ductal adenocarcinoma [10,24].
Future Directions and Emerging Innovations
Improved and Targeted Delivery Systems
Current LNP formulations possess limited tissue-targeting specificity. Selective organ targeting (SORT) strategies involve manipulation of the lipid composition to redirect LNP biodistribution toward the lung, spleen, and lymph nodes. Selective pulmonary delivery via inhaled mRNA-LNP formulations could enable direct mucosal immune induction in the respiratory tract, potentially conferring more durable protection against respiratory pathogens. Engineered extracellular vesicles (EVs) and exosome-based delivery systems are being explored as biomimetic alternatives [14,48].
Artificial Intelligence-Driven Vaccine Design
Integration of artificial intelligence (AI) and machine learning (ML) in mRNA vaccine development is accelerating innovation across multiple design dimensions. ML algorithms including artificial neural networks (ANNs), gradient-boosted trees, and deep learning architectures are being applied to optimise mRNA secondary structures, predict codon optimisation outcomes, identify stability-conferring sequence variants, and design ionisable lipids with enhanced delivery efficiency. AI-guided platforms such as AGILE have demonstrated the ability to screen thousands of lipid candidates in silico, experimentally validate top-ranked hits, and iteratively refine lipid libraries [14].
Multivalent mRNA Vaccines and Cancer Immunotherapy Advances
The dose-flexible, scalable nature of the mRNA platform makes it suited for multivalent vaccines encoding antigens from multiple pathogens within a single formulation. Combination nucleoside-modified mRNA influenza vaccines encoding antigens from 20 different subtypes have demonstrated broad immune coverage without significant immunodominance hierarchies in preclinical studies, representing a potential pathway to universal influenza vaccines [10,52].
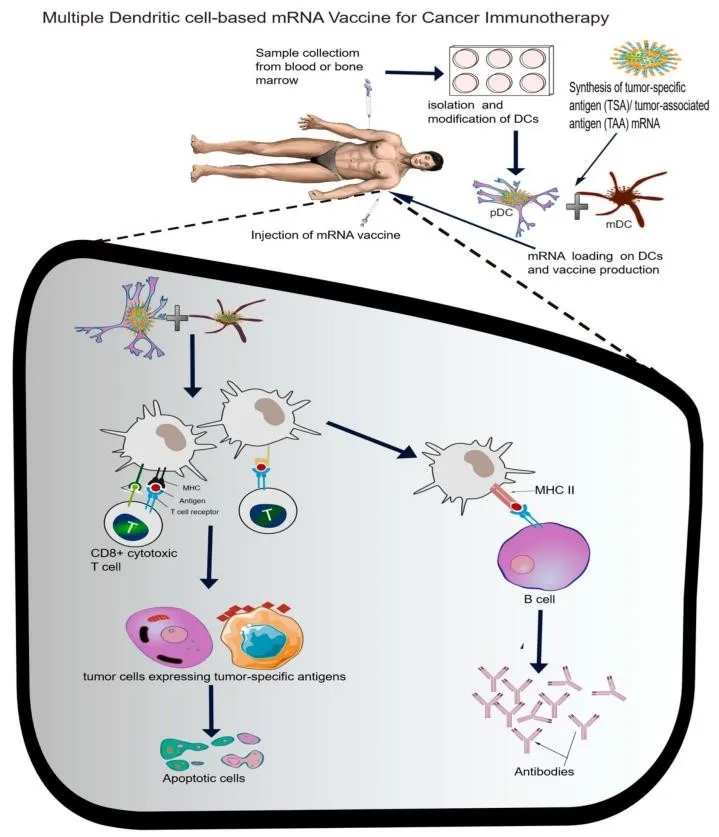
Figure 13. Scheme for multiple dendritic cell (DC)-based mRNA cancer vaccine strategy. Tumour-specific neoantigen mRNA is loaded onto combined myeloid DC (mDC) and plasmacytoid DC (pDC), activating MHC I/II pathways, co-stimulatory molecules, and cytokine secretion to elicit specific CD8+ cytotoxic T cell and antibody responses against tumour cells.
mRNA technology may also enable novel vaccine-therapy combination products co-encoding tumour antigens alongside immunostimulatory payloads (cytokines, co-stimulatory ligands, or checkpoint inhibitor mimetics) to simultaneously activate antigen-specific immunity and relieve immunosuppressive tumour microenvironment signalling [24,47].
CONCLUSION
Messenger RNA vaccine technology has undergone a remarkable transformation from a theoretical concept to a globally deployed public health tool within three decades. The COVID-19 pandemic served as an unprecedented crucible for mRNA vaccine science, compressing development timelines, demonstrating the platform's extraordinary adaptability, and revealing both its remarkable capabilities and its residual limitations. The structural sophistication of contemporary mRNA vaccines—incorporating Cap 1 structures, optimised UTRs, segmented poly(A) tails, nucleoside modifications, and precisely formulated LNP delivery systems—reflects decades of multidisciplinary scientific effort. Looking forward, the trajectory of mRNA technology development is characterised by convergent innovation across delivery, stability, sequence design, and clinical application. The resolution of persistent cold-chain dependencies through lyophilised formulations and thermostable constructs will be foundational to achieving equitable global access. The expanding portfolio of clinical programmes targeting influenza, HIV, RSV, CMV, cancer, and rare genetic diseases demonstrates that COVID-19 vaccines represent only the opening chapter of a far broader mRNA therapeutics revolution. As manufacturing infrastructure matures, regulatory frameworks harmonise, and public confidence in nucleic acid-based medicines is consolidated, mRNA technology is poised to play a defining role in twenty-first century medicine.
Conflicts of Interest: The author declares no conflicts of interest.
Funding: This review received no external funding.
REFERENCES


Afra Shaireen Mohamed Bakkar*, Shivprasad Sanjay Dhage, Messenger RNA Vaccine Platforms in COVID-19: Architecture, Immunological Mechanisms, Delivery Innovations, and Translational Horizons, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 4895-4915. https://doi.org/10.5281/zenodo.20757719
 10.5281/zenodo.20757719
10.5281/zenodo.20757719