
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutical Chemistry, Shivlingeshwar College of Pharmacy, Almala
Pyrrole derivatives are important heterocyclic compounds with significant biological activities. In the present study, pyrrole derivatives were synthesized using microwave-assisted Paal–Knorr synthesis and characterized using FTIR, UV, LC-MS, 1H NMR and 13C NMR studies. Antibacterial activity against Staphylococcus aureus and molecular docking studies were performed.
Heterocyclic compounds represent an important class of organic molecules due to their broad applications in medicinal chemistry and pharmaceutical sciences. Among nitrogen-containing heterocycles, pyrrole has gained considerable attention because of its unique structural characteristics and diverse biological activities. Pyrrole (C₄H₅N) is a five-membered aromatic heterocyclic compound containing one nitrogen atom, where the lone pair of electrons contributes to aromatic stabilization according to Hückel’s rule [1]. The electron-rich nature of pyrrole makes it highly reactive toward electrophilic substitution reactions, especially at C-2 and C-5 positions.
Pyrrole derivatives occur naturally in several biologically important molecules such as heme, chlorophyll, vitamin B12, and cytochromes, indicating their significance in essential physiological functions [2]. Owing to these properties, pyrrole has become an important pharmacophore in medicinal chemistry.
Pyrrole derivatives possess a wide spectrum of biological activities including antimicrobial, anti-inflammatory, antioxidant, antiviral, anticancer, anticonvulsant, and antihypertensive effects [3]. The biological activity of pyrrole compounds mainly depends upon the nature and position of substituents attached to the pyrrole ring. Structural modifications at various positions influence pharmacokinetic properties, receptor binding affinity, and biological selectivity [4].
Several clinically important drugs contain pyrrole or pyrrole-related scaffolds such as Atorvastatin, Sunitinib, Ketorolac, and Tolmetin, demonstrating the pharmaceutical significance of this nucleus [5]. Among various synthetic methods available for pyrrole synthesis, the Paal–Knorr synthesis is one of the most commonly used methods involving cyclization of 1,4-dicarbonyl compounds with primary amines or ammonia [6].
Recently, the development of environmentally friendly synthetic approaches has attracted significant interest in medicinal chemistry research. Microwave-Assisted Organic Synthesis (MAOS) has emerged as an efficient green chemistry technique due to its ability to reduce reaction time, improve yield, minimize solvent use, and decrease energy consumption [7]. Microwave-assisted synthesis of pyrrole derivatives has shown considerable advantages over conventional heating methods by providing rapid and efficient synthesis under eco-friendly conditions.
Molecular docking has also become an important computational approach in drug discovery for predicting ligand–receptor interactions and evaluating binding affinity. It provides useful information regarding molecular interactions and helps in identifying compounds with potential biological activity [8].
Therefore, pyrrole derivatives synthesized using microwave-assisted approaches represent promising candidates for the development of novel biologically active compounds with improved pharmacological potential.
MATERIALS AND METHODS
Pyrrole derivatives were synthesized by microwave-assisted Paal–Knorr synthesis using corresponding amines and diketones. Synthesized compounds were purified and analyzed by TLC and spectroscopic techniques.
Synthesized Compounds
N-(3,4-dimethoxybenzyl)-2,5-dimethylpyrrole
Analytical Results
The synthesized compounds showed acceptable melting point, Rf value and percentage yield (75–76%). Spectral studies confirmed successful synthesis.
Table no.1:-Analytical description of N-(3,4-dimethoxybenzyl)-2,5-dimethylpyrrole
|
Parameter |
Property |
|
Chemical name |
N-(3,4-dimethoxybenzyl)-2,5-dimethylpyrrole |
|
Molecular formula |
C₁₅H₁₉NO₂ |
|
Structure |
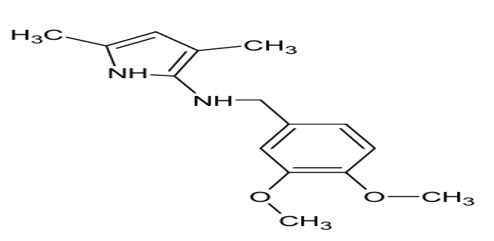
|
|
Molecular weight |
245.32 g/mol |
|
Physical state |
Solid |
|
Appearance |
Off-white to pale yellow crystalline powder* |
|
Odor |
Characteristic |
|
Solubility |
Soluble in methanol, ethanol, chloroform, DMSO; sparingly soluble in water* |
|
Nature |
Aromatic heterocyclic compound |
|
Melting point |
110-115oc |
|
Rf value (TLC) |
0.48 |
|
Microwave irradiation synthesis % yield |
75% |
RESULTS AND DISCUSSION
Spectral Characterization
FTIR confirmed functional groups, UV confirmed aromatic conjugation, LC-MS verified molecular mass, while 1H and 13C NMR confirmed proton and carbon environments.
N-(3,4-dimethoxybenzyl)-2,5-dimethylpyrrol
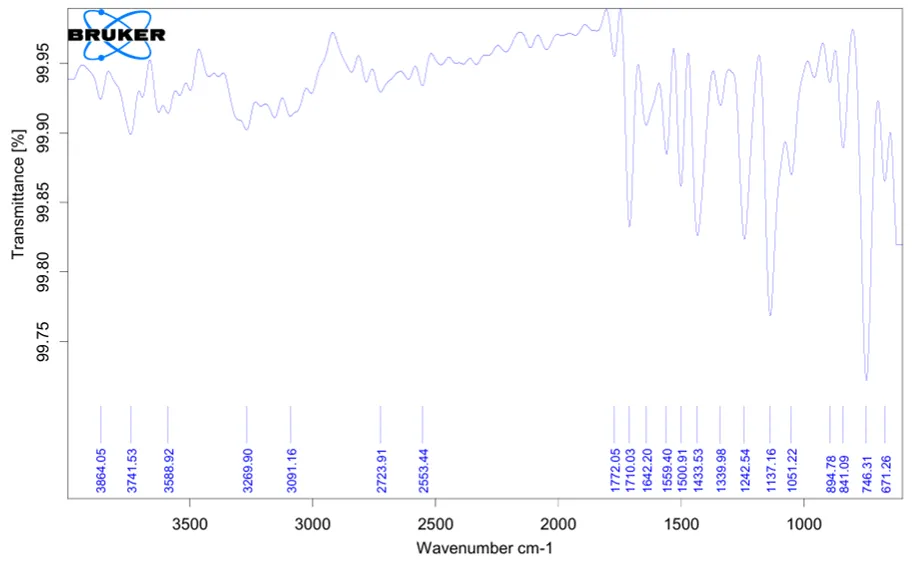
Fig..no.1:- IR spectra of 3,4-dimethoxybenzylamine
Table .no.2:-IR spectra of 3,4-dimethoxybenzylamine interpretation
|
Peak (cm⁻¹) |
Functional Group |
Interpretation |
|
3269 |
N–H stretching |
Indicates presence of primary amine group |
|
3091 |
Aromatic C–H stretching |
Confirms aromatic ring |
|
1559 |
N–H bending |
Confirms amino group |
|
1500 |
Aromatic C=C stretching |
Indicates aromatic structure |
|
1242 |
Ar–O–CH₃ stretching |
Indicates methoxy group |
|
1137 |
C–O stretching |
Confirms ether linkage |
|
1051 |
C–O stretching |
Supports presence of methoxy group |
Discussion:
The FTIR spectrum of the synthesized compound showed characteristic absorption peaks corresponding to the expected functional groups. The peak at 3269 cm⁻¹ confirmed N–H stretching of the primary amine group. Aromatic C–H stretching was observed at 3091 cm⁻¹, while the peak at 1500 cm⁻¹ confirmed aromatic C=C stretching. Peaks at 1242, 1137 and 1051 cm⁻¹ indicated the presence of methoxy and C–O groups. The observed peaks supported the proposed structure of the synthesized compound.
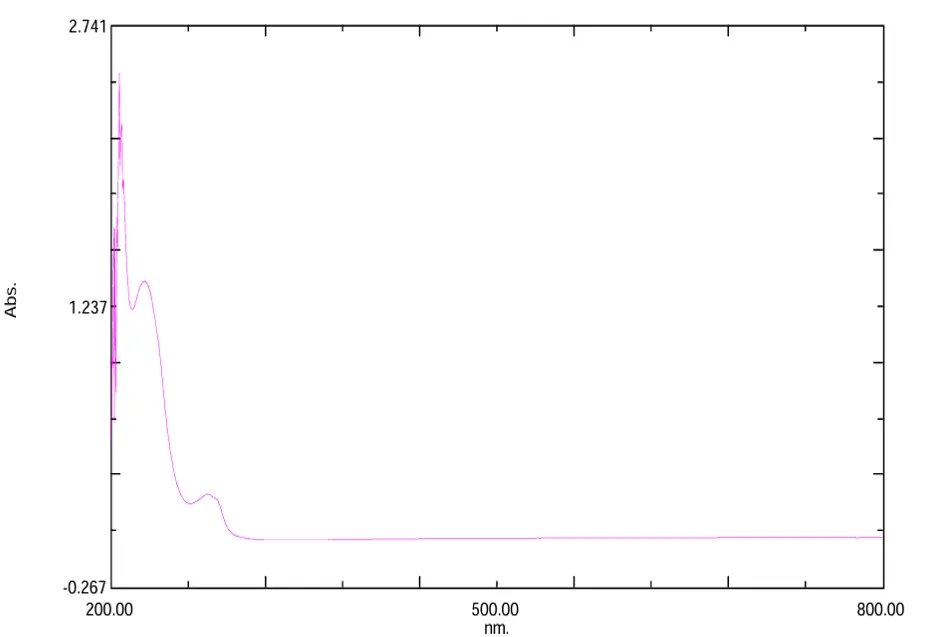
Fig.no2:- UV spectrum of 3,4-dimethoxybenzylamine
Table.no.3:- UV spectrum of 3,4-dimethoxybenzylamine Interpretation
|
Wavelength (nm) |
Assignment |
Interpretation |
|
205–210 |
π→π* transition |
Indicates presence of aromatic ring system |
|
225–240 |
Conjugated absorption band |
Confirms conjugation within the molecule |
|
290–310 |
n→π* transition |
Indicates presence of heteroatom-containing groups |
Interpretation:
The UV–Visible spectrum of the synthesized compound showed characteristic absorption peaks in the ultraviolet region. The absorption peak observed around 205–210 nm was attributed to π→π* electronic transition of the aromatic ring system. A broad absorption band observed around 225–240 nm indicated the presence of conjugation in the molecule. The absorption peak around 290–310 nm was assigned to n→π* electronic transition associated with heteroatom-containing functional groups.
Discussion:
The UV–Visible spectral study of the synthesized compound revealed characteristic absorption bands corresponding to different electronic transitions within the molecule. The observed absorption peaks confirmed the presence of aromatic and conjugated systems in the synthesized compound. The obtained spectral data were found to be in agreement with the proposed molecular structure and indicated successful synthesis of the compound
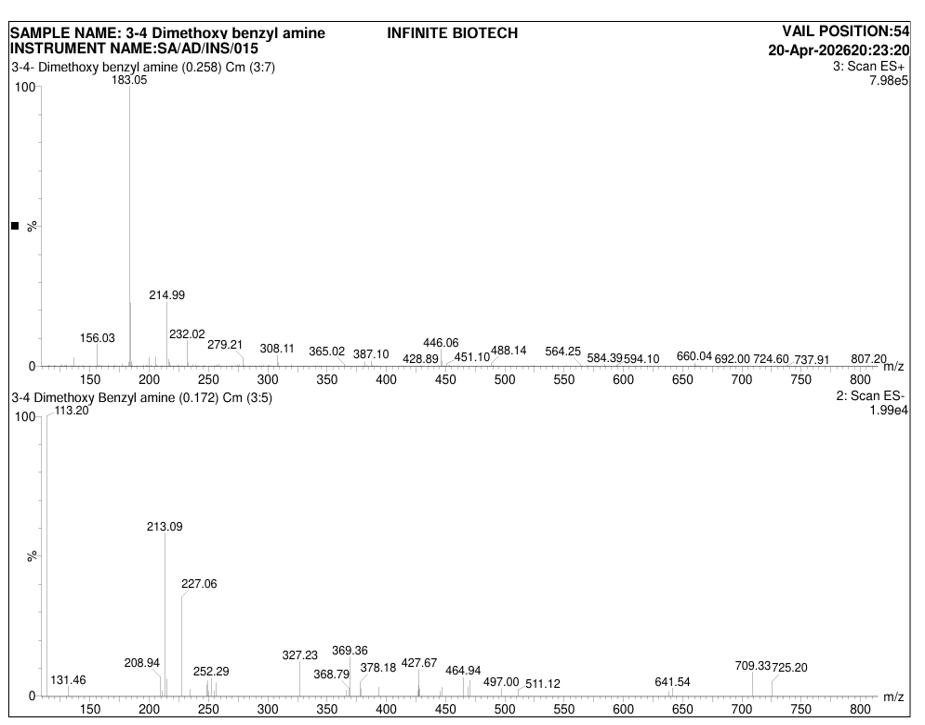
Fig.no.3:-Mass spectrum of 3,4-dimethoxybenzylamine
Table.no.4:-Mass spectrum of 3,4-dimethoxybenzylamine Interpretation
|
m/z Value |
Fragment / Ion |
Interpretation |
|
183.05 [M+H]⁺ |
Molecular ion peak |
Corresponds to protonated molecular ion of synthesized compound |
|
182.05 |
Molecular mass (M) |
Indicates molecular weight of compound |
Interpretation:
The mass spectrum of the synthesized compound showed a prominent molecular ion peak at m/z 183.05 [M+H]⁺, corresponding to the protonated molecular ion of the compound. The observed peak indicated a molecular weight of approximately 182.05 g/mol. The obtained molecular ion peak confirmed the expected molecular mass of the synthesized compound.
Discussion:
The LC–MS spectrum showed a characteristic molecular ion peak corresponding to the synthesized compound. The molecular ion peak observed at m/z 183.05 [M+H]⁺ matched the theoretical molecular weight of the proposed structure. The absence of significant additional peaks suggested good purity of the synthesized compound. The obtained spectral data supported the successful synthesis and structural confirmation of the compound.
Fig.no.4:-Carbon 13 NMR spectrum of 3,4-dimethoxybenzylamine
Table.no.5:-Carbon 13 NMR spectrum of 3,4-dimethoxybenzylamine Interpretation
|
Chemical Shift (δ ppm) |
Carbon Assignment |
Interpretation |
|
148–161 |
Methoxy substituted aromatic carbons |
Indicates aromatic carbons attached to electronegative oxygen atoms |
|
108–129 |
Aromatic carbons |
Confirms presence of aromatic ring carbons |
|
55.5 |
OCH₃ carbons |
Indicates methoxy carbon atoms |
|
~40 |
Benzylic CH₂ carbon |
Indicates methylene carbon attached to aromatic system |
INDEX FREQUENCY PPM HEIGHT
1 19239.9 191.372 3.7 2 17846.8 177.515 5.6 3 16161.4 160.752 4.0 4 15500.7 154.180 1.9 5 15239.0 151.577 4.0 6 15137.6 150.568 5.5 7 14991.8 149.119 6.9 8 14977.4 148.974 4.7 9 14324.3 142.478 5.4 10 13031.9 129.623 2.4 11 12758.7 126.906 5.4 12 12735.8 126.679 4.3 13 12679.4 126.117 2.5 14 12413.9 123.476 3.1 15 12281.9 122.164 5.1 16 11206.1 111.464 4.2 17 11178.7 111.190 6.2 18 10997.9 109.392 2.1 19 10962.8 109.043 3.3 20 10903.3 108.451 3.3 21 5593.2 55.634 5.5 22 5581.0 55.512 5.8 23 4034.5 40.130 24.8 24 4013.2 39.917 75.0 25 3992.6 39.712 149.4 26 3971.2 39.500 174.4 27 3950.6 39.295 148.6 28 3929.2 39.083 74.4 29 3908.6 38.878 24.
Interpretation:
The ¹³C NMR spectrum showed signals corresponding to different carbon environments present in the synthesized compound. Signals in the region of δ 148–161 ppm were assigned to methoxy substituted aromatic carbons due to deshielding caused by oxygen atoms. Signals between δ 108–129 ppm corresponded to aromatic ring carbons. The signal at δ 55.5 ppm indicated methoxy carbon atoms, while the signal around δ 40 ppm was assigned to benzylic CH₂ carbon.
Discussion:
The ¹³C NMR spectrum displayed characteristic carbon signals at expected chemical shift regions. The presence of aromatic carbons, methoxy carbons and benzylic carbon atoms confirmed the structural features of the synthesized compound. The observed chemical shifts were found to be in agreement with the proposed molecular structure and supported successful synthesis of the compound.
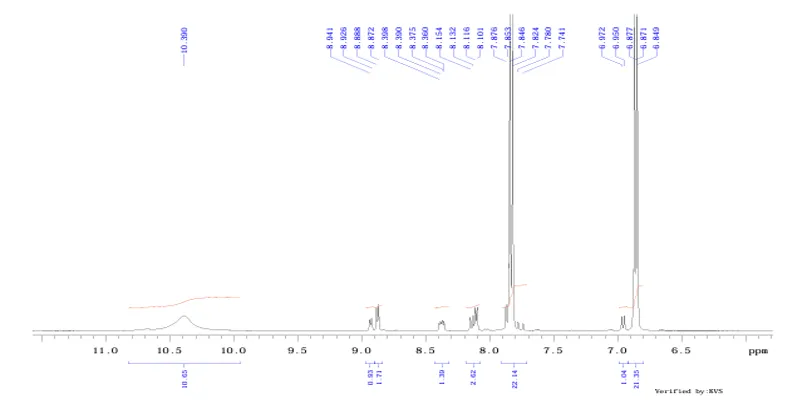
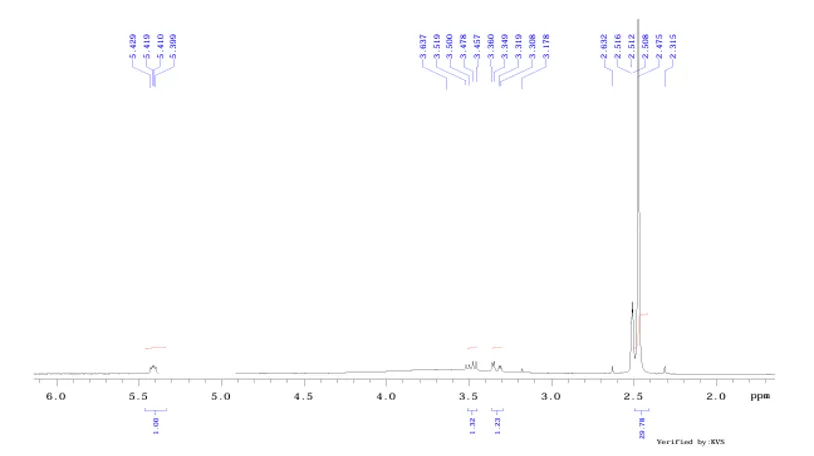
Fig.no.5:-H1 NMR spectroscopy of 3,4-dimethoxybenzylamine
Table.no.6:-H1 NMR spectroscopy of 3,4-dimethoxybenzylamine Interpretation
|
Chemical Shift (δ ppm) |
Proton Assignment |
Interpretation |
|
6.84–6.97 |
Aromatic protons |
Indicates aromatic hydrogen atoms |
|
3.45–3.64 |
CH₂–NH₂ |
Indicates methylene group attached to amino group |
|
~3.8 |
OCH₃ |
Indicates methoxy groups |
|
5.39–5.43 |
NH₂ protons |
Confirms amino protons |
INDEX FREQUENCY PPM HEIGHT
1 4154.4 10.390 6.6 2 3574.8 8.941 4.8 3 3568.7 8.926 5.5 4 3553.5 8.888 10.2 5 3547.2 8.872 11.7 6 3357.9 8.398 3.2 7 3354.7 8.390 4.0 8 3348.4 8.375 4.6 9 3342.5 8.360 4.2 10 3260.1 8.154 5.9 11 3251.5 8.132 6.6 12 3245.2 8.116 10.2 13 3238.8 8.101 10.3 14 3149.1 7.876 11.5 15 3140.0 7.853 28.9 16 3137.1 7.846 140.5 17 3128.3 7.824 154.3 18 3110.7 7.780 3.9 19 3095.0 7.741 3.2 20 2787.4 6.972 6.3 21 2778.6 6.950 6.6 22 2749.8 6.877 26.7 23 2747.1 6.871 147.8 24 2738.3 6.849 147.7 25 2170.7 5.429 2.7 26 2166.6 5.419 3.4 27 2163.1 5.410 3.3 28 2158.7 5.399 3.0 29 1454.2 3.637 1.9 30 1407.0 3.519 3.9 31 1399.2 3.500 3.8 32 1390.4 3.478 5.1 INDEX FREQUENCY PPM HEIGHT 33 1382.3 3.457 5.0 34 1343.5 3.360 4.8 35 1339.1 3.349 5.0 36 1326.8 3.319 3.3 37 1322.4 3.308 3.3 38 1270.6 3.178 2.1 39 1052.5 2.632 3.3 40 1006.0 2.516 19.7 41 1004.3 2.512 27.9 42 1002.6 2.508 25.1 43 989.6 2.475 570.1 44 925.6 2.315 3.2 45 -0.0 -0.000 25.
Discussion:
The ¹H NMR spectrum showed characteristic proton signals corresponding to the synthesized compound. Signals in the region of δ 6.84–6.97 ppm indicated aromatic protons. The signal at δ 3.45–3.64 ppm confirmed CH₂ attached to the amino group. The signal observed around δ 3.8 ppm indicated methoxy groups, while signals at δ 5.39–5.43 ppm represented NH₂ protons. The obtained signals were in agreement with the expected structure.
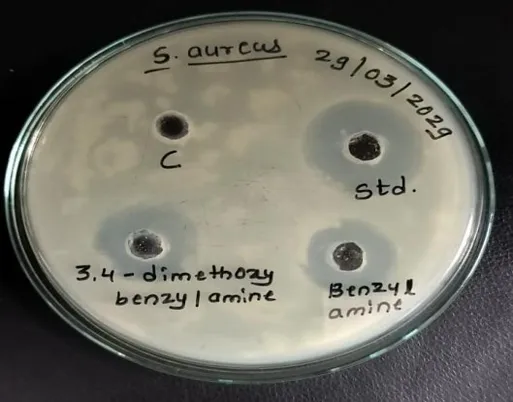
Antibacterial Activity
The compounds showed moderate antibacterial activity against Staphylococcus aureus. 3,4-dimethoxybenzyl derivative showed highest inhibition (19 mm) among synthesized compounds
Table No.7:- Antibacterial activity of test compound against S. aureus
|
SR. NO |
SAMPLES |
ZONE IN DIAMETER (mm) |
|
1 |
Control |
00 |
|
2 |
Standard (Streptomycin) |
26 |
|
3 |
3,4-dimethoxybenzyl |
19 |
|
4 |
Benzyl amine |
15 |
|
5 |
Phenyl amine |
18 |
Image Activity:
Conclusion of the study:
The antibacterial activity against Staphylococcus aureus was evaluated using the zone of inhibition method, where the control showed no inhibition (0 mm), confirming the reliability of the assay. The standard antibiotic, Streptomycin, exhibited a strong inhibition zone of 26 mm, indicating high antibacterial efficacy. Among the test compounds, 3,4-dimethoxybenzyl showed the highest activity with a zone of 19 mm, followed by phenyl amine (18 mm) and benzyl amine (15 mm). Although all test compounds demonstrated lower activity compared to the standard Streptomycin, their measurable inhibition zones indicate moderate antibacterial potential against Staphylococcus aureus. Overall, 3,4-dimethoxybenzyl was found to be the most effective among the tested compounds.
N-(3,4-dimethoxybenzyl)-2,5-dimethylpyrrole
Molecular Docking and ADME
Docking score ranged from −3.94 to −4.72 kcal/mol. Compound A.J.3 showed strongest binding affinity with MMGBSA value of −38.86 kcal/mol and passed Lipinski parameters.
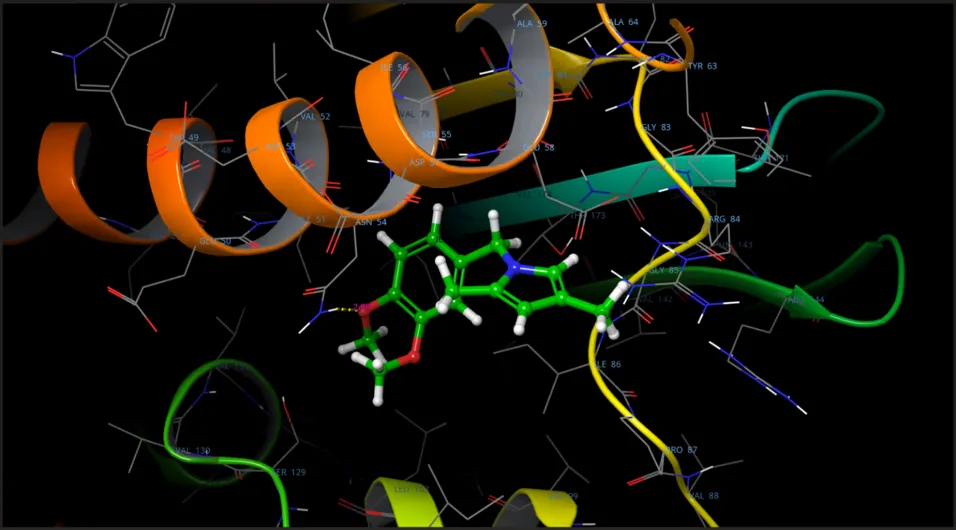
Fig.no.6:- 3D binding pose of N-(3,4-dimethoxybenzyl)-2,5-dimethylpyrrole
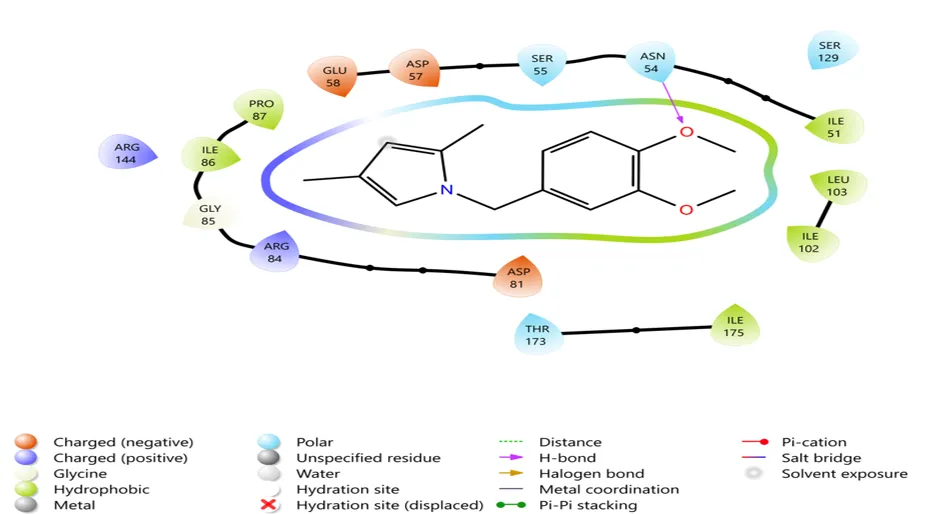
Fig.no.7:- 2D interaction diagram of N-(3,4-dimethoxybenzyl)-2,5-dimethylpyrro
Table no.8:-Molecular Docking and MM-GBSA Results
|
Compound Code |
Docking Score (kcal/mol) |
MMGBSA Binding Energy (kcal/mol) |
Human Oral Absorption (%) |
Lipinski Rule |
|
A.J.3 |
-4.72 |
-38.86 |
100 |
Passed |
Interpretation
The molecular docking results indicated that the synthesized pyrrole derivative exhibited favorable interaction with the target receptor protein. Compound A.J.3 showed a docking score of −4.72 kcal/mol, indicating good receptor binding affinity and stronger interaction with the active site residues.
The MMGBSA binding energy value further supported the docking result. Compound A.J.3 displayed a ΔG bind value of −38.86 kcal/mol, suggesting higher binding stability with the receptor protein.
The ADME study predicted that compound A.J.3 possesses 100% human oral absorption and satisfied Lipinski's Rule of Five, indicating favorable drug-likeness properties and supporting its potential as a bioactive molecule for further investigation.
CONCLUSION
The present molecular docking and ADME study demonstrated that the synthesized pyrrole derivatives possess promising receptor binding characteristics and acceptable drug-likeness properties. Compound A.J.3 exhibited comparatively better binding affinity and stability among the tested compounds and may serve as a potential lead compound for further biological investigation.
REFERENCES


Jadhav Asmita, Dr. Malpani Suraj, Dr. Dharashive Vishweshwar, Microwave Assisted Synthesis and Evaluation of Pyrrole Derivative, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 7, 1607-1618. https://doi.org/10.5281/zenodo.21261458
 10.5281/zenodo.21261458
10.5281/zenodo.21261458