
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1Research Scholar, Guru Nanak College of Pharmaceutical Sciences, Dehradun
2Professor, Guru Nanak College of Pharmaceutical Sciences, Dehradun
The New Drug Application (NDA) is the formal regulatory submission through which a pharmaceutical sponsor seeks permission from the U.S. Food and Drug Administration (FDA) to market a new drug for human use. This article provides a comprehensive overview of the regulatory approval process for an NDA, beginning with the essential preclinical and clinical development phases that precede submission. Preclinical research (3–6 years) evaluates safety and toxicity in animal models, while the Investigational New Drug (IND) application serves as the gateway to human trials. Three sequential clinical trial phases follow: Phase 1 (safety and dosage, 20–80 participants), Phase 2 (efficacy and side effects, several dozen to 300 participants), and Phase 3 (confirmatory evidence, several hundred to 3,000+ participants). Following successful trial completion, sponsors may request a Pre-NDA meeting with the FDA to align expectations. The NDA itself comprises key components: comprehensive clinical data (including integrated summaries of safety and efficacy), proposed labeling (package insert), manufacturing information (Chemistry, Manufacturing, and Controls), and supportive pharmacokinetic/pharmacodynamic studies. Submission is mandated in the Electronic Common Technical Document (eCTD) format, an XML-based structured system organized into five modules. The FDA then conducts a five-step review process: filing determination (within 60 days), formal review period (6–10 months for standard review, 6 months for priority review), multidisciplinary team evaluation (medical officers, statisticians, pharmacologists, chemists), on-site inspections (clinical sites and manufacturing facilities), and final action package leading to an approval letter, complete response letter, or non-approval letter. Post-approval, Phase 4 studies monitor long-term safety. Understanding this rigorous, multi-year process underscores the NDA’s critical role as a regulatory firewall protecting public health while enabling access to novel therapies.
The Regulatory Approval Process of a New Drug Application (NDA)
The path from a promising molecular hypothesis to a prescription medicine available at a local pharmacy is one of the most arduous, expensive, and highly regulated journeys in modern science. At the heart of this journey’s final stage lies a critical document: the New Drug Application (NDA). An NDA is the formal, comprehensive proposal that a pharmaceutical sponsor submits to a regulatory authority—most notably the U.S. Food and Drug Administration (FDA)—seeking official permission to market and distribute a new drug for human use. This application is not a simple request; it is a veritable archive of scientific evidence, containing thousands of pages detailing everything from a drug’s chemical structure and manufacturing process to the raw data from all animal studies and human clinical trials. The role and importance of the NDA process cannot be overstated. It serves as the regulatory firewall between experimental therapy and public health, ensuring that any drug entering the marketplace has been rigorously proven to be both safe and effective for its intended use. Beyond protecting patients, the NDA process upholds the integrity of the medical profession and fosters public trust in the pharmaceutical industry. For a sponsor, successful NDA approval is the culmination of a decade or more of investment, the gateway to revenue, and the only legal pathway to bringing life-saving or life-improving therapies to those who need them. However, to truly understand the gravity of the NDA, one must first appreciate the immense scientific and regulatory journey that precedes its submission—a multi-phase lifecycle designed to winnow out failures long before they ever reach the FDA’s review desk[1][2]. The drug development lifecycle is a methodical, stepwise process that begins long before a single human volunteer takes a dose. The initial stage, preclinical research, typically spans three to six years and is conducted primarily in laboratories and on animal models. Long before a molecule becomes a candidate drug, it must be identified, synthesized, and characterized. Researchers explore its pharmacodynamics (what the drug does to the body, e.g., binding to a specific receptor) and pharmacokinetics (what the body does to the drug, e.g., absorption, distribution, metabolism, and excretion).
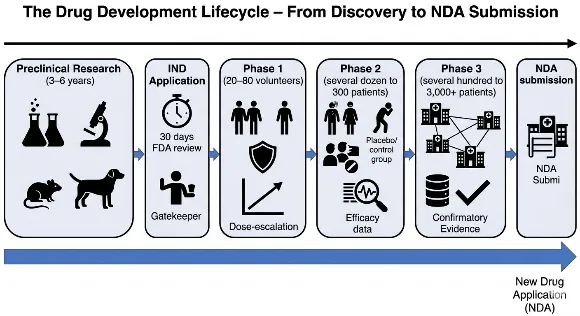
Fig: 1 Drug Development Lifecycle – From Discovery to NDA Submission
However, the core mandate of preclinical research is safety. Through rigorous in vitro (test tube or cell culture) studies and in vivo (living organism) studies in at least two animal species (commonly rodents and non-rodents like dogs or monkeys), sponsors must gather initial data on toxicity, dosing thresholds, and potential side effects. These studies investigate acute toxicity (single high dose), subacute and chronic toxicity (repeated doses over weeks or months), genotoxicity (potential to cause DNA damage), carcinogenicity (potential to cause cancer), and reproductive toxicity (effects on fertility and fetal development)[3][4]. If a candidate drug shows unacceptable toxicity or no therapeutic promise at this stage, it is terminated, saving the immense resources that would be required for human trials. Only those molecules that demonstrate a reasonable safety profile and evidence of potential efficacy in animals advance to the next critical juncture: the Investigational New Drug (IND) application.
The IND application is the formal gateway that, once approved, allows a sponsor to move from laboratory and animal testing into human clinical trials. Think of it as a request for an exemption from the federal law that prohibits shipping an unapproved drug across state lines. The IND is a substantial submission that includes three major components: the animal pharmacology and toxicology data from the preclinical phase, the chemistry and manufacturing information (demonstrating the drug can be consistently produced with purity and potency), and a detailed protocol for the first human trial (Phase 1). Crucially, the IND must include any prior human experience (e.g., from foreign studies) and the investigator’s brochure—a document summarizing everything known about the drug for the clinical investigators. When the FDA receives an IND, a 30-day review clock begins. If the agency does not issue a clinical hold (a pause due to safety concerns or protocol flaws) within that period, the sponsor may proceed with Phase 1 trials. The IND is not a one-time approval; it remains active throughout the clinical development process, requiring annual reports and amendments for protocol changes or new safety findings. It is the living license to explore the drug in humans[5][6]. With an active IND, the sponsor initiates the three sequential phases of clinical trials, each designed to answer specific questions. Phase 1, typically lasting several months to a year, is the first human exposure and is primarily concerned with safety and dosage. This phase involves a small number of participants—usually 20 to 80 healthy volunteers (though for oncology or other life-threatening diseases, volunteers are often actual patients)[7]. Phase 1 trials are often conducted in specialized clinical pharmacology units. The goals are manifold: determine the most common adverse reactions, understand how the drug is metabolized and excreted, identify a safe and tolerable dose range, and observe any preliminary evidence of efficacy. Researchers use a process called dose-escalation, starting with a very low, sub-therapeutic dose and carefully increasing it in subsequent groups of volunteers while monitoring for toxicities. This phase is deliberately cautious; serious adverse events can halt development immediately. By the end of Phase 1, the sponsor should have a reasonably safe maximum tolerated dose (MTD) and a recommended dose for the next phase[8]. If Phase 1 is successful, the sponsor moves to Phase 2, which focuses on efficacy and side effects. This phase enrolls several dozen to approximately 300 participants, who are now patients with the target disease or condition. Phase 2 trials are typically controlled and randomized, meaning one group receives the experimental drug while a control group receives a placebo or standard treatment. The primary objective is to assess whether the drug has any biological activity or clinical benefit against the disease. Does it shrink tumors[8] Lower blood pressure? Reduce infection rates? Simultaneously, Phase 2 continues to evaluate short-term safety and side effects in a larger, more diverse population than Phase 1. These studies often use different dosing regimens (e.g., once daily vs. twice daily) to optimize the balance between efficacy and tolerability. A Phase 2 trial is often a "go/no-go" decision point: if the drug fails to show any signal of efficacy or demonstrates unacceptable toxicity, it will likely be abandoned. Approximately 70% of drugs fail between Phase 1 and Phase 2. For those that pass, a more definitive and expensive Phase 3 is launched[9][10]. Phase 3 trials represent the pivotal, confirmatory evidence of a drug’s value. These are large-scale, randomized, double-blind, controlled trials involving typically several hundred to 3,000 or more patients at multiple clinical sites across the country or even globally. The design of Phase 3 trials is rigorous, often comparing the new drug head-to-head against a placebo, a standard-of-care treatment, or both. The primary goal is to confirm the efficacy observed in Phase 2 on a broader scale and to accumulate the comprehensive safety database necessary to detect less common adverse events—those that might occur in only 1 in 500 or 1 in 1,000 patients. Phase 3 trials also explore different populations (e.g., various age groups, disease severities, or patients with kidney or liver impairment) and may study drug interactions. These studies typically take one to four years and cost hundreds of millions of dollars. Successful completion of Phase 3 yields the definitive evidence that the drug’s benefits outweigh its risks for a defined indication, and it generates the statistical datasets that form the core of the NDA. It is important to note that failure at this stage is not rare; drugs can fail due to a lack of superior efficacy over existing treatments, unexpected toxicity in a large population, or even commercial reasons related to the changing market landscape[11]. Only after all three phases of clinical trials are complete and the data have been meticulously analyzed does a sponsor begin to assemble the NDA. The NDA itself is organized into a standardized technical structure known as the Common Technical Document (CTD), which is accepted by the FDA and many other global regulators. The CTD is divided into five modules. Module 1 contains administrative information, proposed labeling, and prescribing information. Module 2 provides high-level summaries and overviews of the quality, nonclinical, and clinical data. Module 3 details the chemistry, manufacturing, and controls (CMC)—how the drug substance is made and how the final drug product (tablet, injection, etc.) is formulated, tested for stability, and packaged[12][13]. Module 4 presents all the preclinical animal study reports. Module 5, the largest and most scrutinized, contains all the clinical study reports, including the full protocols, statistical analysis plans, patient-level data listings, and integrated summaries of safety and efficacy from the Phase 1, 2, and 3 trials. Upon submission, the FDA has 60 days to conduct a preliminary review to ensure the NDA is sufficiently complete to permit a full review. If accepted for filing, the FDA assigns a review classification: Standard (10-month review goal) or Priority (6-month review goal for drugs that offer significant therapeutic advances). The review involves a multidisciplinary team of medical officers, pharmacologists, chemists, statisticians, and others who scrutinize every data point. They may request advisory committee meetings with outside experts, issue information requests, or conduct site inspections of clinical trial centers. At the end of the review, the FDA issues an action letter: an approval letter, a complete response letter (denying approval and listing deficiencies that must be corrected), or rarely, a non-approval letter. Approval allows the drug to enter the U.S. market, but it does not end the drug’s lifecycle. The sponsor must continue to submit periodic safety reports, and Phase 4 (post-marketing) studies are often required to monitor long-term safety in the general population. In summary, the NDA is not merely a regulatory hurdle; it is the logical and necessary capstone of a rigorous, multi-year scientific process that transforms a laboratory curiosity into a trusted medicine, safeguarding the public one application at a time[14].
Preparing and Submitting the NDA
Once a sponsor has successfully navigated the treacherous waters of preclinical research and three phases of clinical trials, a natural and critical question arises: is the accumulated evidence sufficient to convince the FDA that the drug is safe and effective? Rather than submitting the New Drug Application (NDA) in a vacuum, astute sponsors avail themselves of one of the most valuable opportunities in the regulatory calendar: the Pre-NDA meeting with the FDA. This meeting, typically requested several months before the planned NDA submission, is a formal, face-to-face (or virtual) dialogue between the sponsor and the agency’s review division. Its purpose is not to obtain preliminary approval but to ensure that the forthcoming NDA is complete, well-organized, and likely to be accepted for filing. During the Pre-NDA meeting, the sponsor presents a high-level summary of the pivotal efficacy and safety data, the proposed labeling, and any lingering concerns—such as unresolved statistical issues or missing data from a small number of patients[15][16]. The FDA team, including medical officers, statisticians, and pharmacologists, provides critical feedback. They may confirm that the proposed clinical studies are adequate to support approval, or they may request additional analyses, clarify what safety data should be pooled, or flag deficiencies that would otherwise trigger a complete response letter. Perhaps most importantly, the agency will indicate whether it intends to hold an advisory committee meeting—a public session where outside experts debate the drug’s risks and benefits. The Pre-NDA meeting is not legally required, but skipping it is considered unwise. Sponsors who attend these meetings dramatically reduce the risk of receiving a “refuse to file” letter, which would delay submission by months. Moreover, the meeting fosters transparency and alignment, transforming the NDA process from an adversarial audit into a collaborative scientific endeavor. By the end of this meeting, both parties share a clear roadmap for the NDA’s structure, content, and anticipated review timeline[17][18]. With the insights from the Pre-NDA meeting in hand, the sponsor begins the monumental task of assembling the NDA itself. The application is far more than a simple collection of study reports; it is an integrated, self-contained argument for approval, and its key components can be grouped into several essential categories. The first and most substantial component is the clinical data. This includes the complete results from all Phase 1, 2, and 3 trials, presented in standardized tabular and narrative formats. However, raw data alone are insufficient. The sponsor must provide integrated summaries of safety and efficacy, which combine data across multiple trials to detect rare adverse events or subpopulation effects. For example, a drug studied in 2,000 patients across three Phase 3 trials might show a low overall rate of liver enzyme elevations, but when the data are pooled, a signal might emerge among elderly patients with renal impairment. The integrated summary of safety (ISS) and integrated summary of efficacy (ISE) are the heart of the NDA’s clinical argument. Additionally, the sponsor must submit patient-level data listings, case report forms for any patient who died or dropped out due to an adverse event, and statistical analysis plans that demonstrate the analyses were pre-specified, not data-mined after the fact. The clinical data component also includes any supportive studies, such as dose-response trials or comparative effectiveness studies against an active comparator. Without comprehensive, transparent, and well-analyzed clinical data, the NDA cannot even be considered for filing[19][20]. A second critical component is the labeling information, specifically the proposed package insert. The package insert—often called the “label”—is the legally binding document that accompanies every dispensed prescription. It is not a marketing brochure but a meticulously worded scientific summary that tells healthcare providers everything they need to know: the drug’s approved indications (the diseases or conditions it treats), dosage and administration instructions, contraindications (when the drug must not be used), warnings and precautions, adverse reactions, drug interactions, use in specific populations (pregnancy, lactation, pediatrics, geriatrics), and clinical study results. The sponsor drafts the proposed label based on the trial data, but the FDA has final authority over every word. During the NDA review, intense negotiations occur over the wording of the “Warnings and Precautions” section. For instance, a drug that causes a 1% incidence of serious liver injury might require a “boxed warning”—the FDA’s strongest caution. The sponsor may wish to claim efficacy in a broad patient population, but the FDA may restrict the indication to a narrower subgroup based on the trial’s inclusion criteria. The proposed label also includes the “Patient Information” or “Medication Guide” that must be given to patients[21].
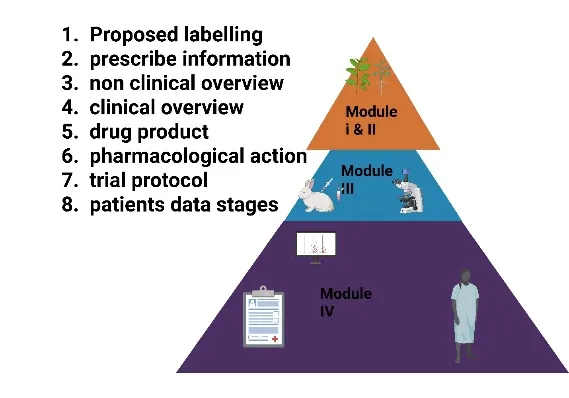
Fig: 2 Key Components of an NDA – The eCTD Modular Structure
Submitting a well-justified, evidence-based proposed label is not a mere formality; it demonstrates that the sponsor understands the drug’s risk-benefit profile and can communicate it responsibly. A poorly drafted label that overstates benefits or downplays risks can derail the entire application[22]. The third essential component is manufacturing information, often referred to as Chemistry, Manufacturing, and Controls (CMC). This section details how the drug substance (the active pharmaceutical ingredient) is synthesized, purified, and characterized, and how the final drug product (the tablet, capsule, injectable solution, etc.) is formulated, filled, packaged, and labeled. The CMC section must demonstrate that every batch of the drug is produced consistently, with the same identity, strength, quality, purity, and potency. This requires submitting detailed standard operating procedures, specifications for raw materials, in-process testing protocols, and final product release tests (e.g., dissolution testing for tablets, sterility testing for injectables). Stability data are paramount: the sponsor must show that the drug remains within specifications over its proposed shelf life under various storage conditions (e.g., 25°C/60% relative humidity and 40°C/75% relative humidity). For biologics or complex small molecules, the manufacturing information can run thousands of pages, including validation reports for every piece of equipment and every analytical method. The FDA’s Office of Pharmaceutical Quality reviews this section not on paper alone; they may conduct a pre-approval inspection of the manufacturing facility to ensure that the written procedures match real-world practice. Any deficiency in manufacturing—such as impurity levels exceeding a limit, lack of sterility assurance, or inadequate stability data—can lead to a complete response letter, even if the clinical data are perfect. Thus, successful NDA sponsors begin preparing the CMC section years in advance, during the preclinical and Phase 1 stages[23][24]. Beyond clinical data, labeling, and manufacturing, the NDA must include other relevant studies that inform the drug’s safe and effective use. Prominent among these are pharmacokinetic (PK) and pharmacodynamic (PD) studies. PK studies describe what the body does to the drug: how quickly it is absorbed (Cmax, Tmax), how widely it distributes into tissues (volume of distribution), how it is metabolized (which liver enzymes are involved), and how it is excreted (renal or biliary). PK studies are often conducted in healthy volunteers during Phase 1, but they are also performed in special populations: patients with kidney or liver impairment (since impaired clearance can cause toxic accumulation), the elderly, children, and pregnant women. The NDA must include a dedicated clinical pharmacology summary that integrates all PK data, identifies any need for dose adjustments, and flags potential drug-drug interactions. For example, if a drug is metabolized by the CYP3A4 liver enzyme, the sponsor must study what happens when the drug is co-administered with a strong CYP3A4 inhibitor (like ketoconazole) or inducer (like rifampin). PD studies, in contrast, describe what the drug does to the body, often at a molecular or physiological level. They may include biomarker assessments (e.g., receptor occupancy measured by PET imaging) or early efficacy signals (e.g., reduction in LDL cholesterol). When combined, PK/PD modeling can identify the exposure-response relationship, enabling the sponsor to propose a dosing regimen that maximizes efficacy while minimizing toxicity. These studies are not secondary; they are core to the NDA’s scientific validity. A drug that works beautifully in clinical trials but has unpredictable PK in patients with renal failure cannot be approved without a clear dosing recommendation[25]. Having gathered all these components, the sponsor faces one final, formidable challenge: formatting the NDA for submission. Gone are the days of shipping pallets of paper documents weighing hundreds of pounds. Today, the mandatory standard is the Electronic Common Technical Document, or eCTD. The eCTD is a structured, XML-based electronic format that organizes the NDA into a hierarchical set of modules, folders, and files. The backbone of the eCTD is the Common Technical Document (CTD), an internationally harmonized specification agreed upon by the FDA, the European Medicines Agency, Japan’s PMDA, and other regulators. The CTD is divided into five modules. Module 1 is region-specific and contains administrative information, proposed labeling, and the prescribing information in FDA-specified format. Module 2 contains the high-level summaries: the quality overall summary, the nonclinical overview, the clinical overview, and the integrated summaries of safety and efficacy. Module 3 is the quality (CMC) section, organized into drug substance and drug product subsections. Module 4 is the nonclinical study reports (pharmacology, toxicology). Module 5 is the clinical study reports, including the integrated summaries, individual study reports, and patient-level data. The eCTD format offers immense advantages: it allows reviewers to click on hyperlinks to navigate instantly from a summary table to the raw data, it supports electronic signatures and time-stamped document tracking, and it enables the FDA to run automated validations for completeness and technical compliance. Sponsors must use specialized software to generate the eCTD, ensuring that every file is named correctly, that the table of contents is accurate, and that no viruses or corrupted files are present. The FDA requires that the eCTD be submitted on a compact disc, a digital versatile disc, or via the FDA’s electronic submission gateway. The submission must pass a technical validation within 24 hours; if it fails, the FDA may refuse to file the application. Therefore, most sponsors conduct several mock eCTD submissions or “pre-submission readiness checks” to avoid embarrassing and costly rejections. The transition to eCTD has dramatically streamlined the review process, reducing the average review time from 24 months (in the paper era) to 10 months for standard NDAs and 6 months for priority NDAs. However, it has also raised the bar for sponsors, requiring expertise in regulatory informatics as well as drug development. Ultimately, the eCTD-formatted NDA—complete with clinical data, labeling, manufacturing details, and PK/PD studies—represents the final, packaged argument for a new medicine. When that submission is uploaded and the FDA issues its acknowledgment letter, the decade-long journey from lab bench to patient bedside enters its final chapter, awaiting the agency’s verdict[26].
FDA NDA Review Process: A Step-by-Step Breakdown
Following the submission of an electronic Common Technical Document (eCTD), the New Drug Application (NDA) enters a structured, multi-step review process that determines whether a new medicine will reach patients. The first critical juncture is Step 1: the filing determination, which occurs within the first 60 days of receipt. During this period, the FDA conducts a preliminary, gatekeeping review to assess whether the NDA is sufficiently complete and well-organized to warrant a full scientific evaluation. The agency checks for basic administrative compliance—are all five eCTD modules present? Is the integrated summary of safety legible? Have all required patient-level data listings been provided? If the application meets these threshold criteria, the FDA issues an “Acceptance for Filing,” and the review clock officially starts. However, if the NDA has major omissions—such as missing clinical study reports, uninterpretable datasets, or inadequate manufacturing information—the agency will issue a “Refusal to File” (RTF) letter. An RTF is not a final denial of approval; it is a rejection of the application’s format or completeness, forcing the sponsor to correct deficiencies and resubmit, often delaying the process by many months. Fortunately, through the Pre-NDA meeting and careful preparation, most major NDAs are accepted for filing[27]. Once filed, Step 2, the formal review period, begins. For a standard NDA, the FDA has 10 months from the filing date to complete its review, though this can extend to 12 months if major amendments are submitted. For drugs that address an unmet medical need or offer a significant therapeutic advance over existing treatments, the sponsor may request—and the FDA may grant—Priority Review, which shortens the review period to 6 months. Priority Review does not change the approval standards (safety and efficacy remain paramount), but it allocates additional agency resources and faster internal timelines. During these 6 to 10 months, Step 3 unfolds: a multidisciplinary review team evaluates every facet of the application. This team includes medical officers (physicians) who scrutinize the clinical data, assessing whether the trials were well-designed, whether the patient populations were appropriate, and whether the observed benefits convincingly outweigh the risks. They work alongside statisticians who independently re-analyze the raw data to verify the sponsor’s statistical claims, checking for p-hacking, missing data biases, or inappropriate subgroup analyses. Pharmacologists focus on Module 4, reviewing the animal toxicology studies to ensure that the human safety data align with the nonclinical findings, and that any human-specific toxicities (e.g., a rare allergic reaction not seen in animals) are properly addressed. Chemists, within the Office of Pharmaceutical Quality, examine Module 3’s Chemistry, Manufacturing, and Controls (CMC) data to verify that the drug product is consistently produced, stable, and free from contaminants. These four disciplines do not work in silos; they meet regularly in review team meetings, sharing concerns and cross-referencing findings. A medical officer might ask the chemist whether an observed impurity could explain a rare adverse event; the pharmacologist might ask the statistician to re-analyze safety data by dose level. Running parallel to the scientific review is Step 4: on-site inspections. The FDA’s Office of Scientific Investigations sends inspectors to selected clinical study sites that conducted pivotal Phase 3 trials. These inspectors verify that the patient consent forms were properly signed, that adverse events were accurately reported, that the study drug was stored and dispensed correctly, and that no data fabrication occurred. Simultaneously, the Office of Pharmaceutical Quality’s inspectors visit the manufacturing facilities—the sites where the drug substance is synthesized and where the final product is made, packaged, and labeled. They examine equipment calibration logs, cleaning procedures, batch records, and quality control testing labs. Any serious violations, such as evidence of fraud at a clinical site or unsanitary conditions at a factory, can trigger a complete response letter regardless of the drug’s clinical merits. Finally, in Step 5, all findings converge into the “Action Package”—a comprehensive document that includes the review team’s integrated assessment, the inspection reports, the proposed labeling, and a final recommendation to the FDA’s division director. The Action Package may also include briefing materials for an advisory committee meeting if external expert input is needed. The director then issues the final decision: an Approval Letter (allowing marketing), a Complete Response Letter (explaining deficiencies that must be corrected before approval), or rarely, a Non-Approval Letter (for serious, uncorrectable issues). This five-step process, spanning 6 to 12 months, represents the last regulatory mile—a rigorous yet fair examination that honors the decade of work behind the NDA and protects the public health one application at a time[27][28].
CONCLUSION
The journey of a new drug from a molecular hypothesis to a prescription medicine available at the pharmacy counter is nothing short of extraordinary. As this article has detailed, the New Drug Application (NDA) is not an isolated document but the culminating archive of a decade or more of painstaking research, billions of dollars in investment, and the collective efforts of thousands of scientists, clinicians, and regulatory professionals. From the earliest preclinical toxicity studies in animal models to the large-scale, randomized, double-blind Phase 3 trials involving thousands of patients, every step of the drug development lifecycle is designed to answer one fundamental question: does the benefits of this drug outweigh its risks for the intended patient population. The NDA process embodies a careful balance between two competing public health imperatives: the urgent need to bring life-saving and life-improving therapies to patients, and the equally critical obligation to ensure that no unsafe or ineffective drug reaches the market. Mechanisms such as the Pre-NDA meeting, the multidisciplinary review team, on-site inspections of clinical and manufacturing facilities, and the rigorous eCTD formatting standards all serve to uphold this balance. The FDA’s step-by-step review—from the initial 60-day filing determination through the formal 6- to 10-month review period to the final action package—provides a transparent, science-driven, and legally robust framework for decision-making. Whether a drug receives a standard review (10 months) or a priority review (6 months) for significant therapeutic advances, the approval standards remain unchanged: safety and efficacy above all else.Importantly, NDA approval does not mark the end of regulatory oversight. Phase 4 post-marketing studies, periodic safety reports, and the FDA’s ongoing surveillance system ensure that even rare adverse events are detected and addressed after the drug enters general use. This life-cycle approach reflects the understanding that a drug’s risk-benefit profile can evolve as it is used in broader, more diverse populations than those studied in clinical trials.
REFERENCES

Md. Firoz Ansari*, Mohit Gupta, Regulatory Approval Process Of NDA: A Review, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 5, 7108-7118. https://doi.org/10.5281/zenodo.20404706
 10.5281/zenodo.20404706
10.5281/zenodo.20404706