
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Guru Nanak College of Pharmaceutical Science, Dehradun, Uttarakhand, India.
Biosimilars have emerged as a transformative approach to improving access to affordable biologic therapies while reducing healthcare expenditures worldwide. India, being one of the largest producers of pharmaceutical products, has played a significant role in the development and commercialization of biosimilars, commonly referred to as “similar biologics.” The regulatory framework governing biosimilars in India has evolved considerably over the past decade through the collaborative efforts of the Central Drugs Standard Control Organization (CDSCO) and the Department of Biotechnology (DBT). The introduction of the “Guidelines on Similar Biologics” in 2012 and their subsequent revisions aimed to establish a scientifically robust pathway for demonstrating biosimilarity in terms of quality, safety, and efficacy. Despite these advancements, several regulatory challenges persist, including limited transparency in approval procedures, inadequate pharmacovigilance systems, lack of clear interchangeability guidelines, variability in clinical trial requirements, and insufficient harmonization with international regulatory standards such as those of the USFDA and EMA. This review critically examines the current regulatory framework for biosimilar approval in India, including preclinical, clinical, manufacturing, and post-marketing requirements. Furthermore, it highlights existing regulatory gaps and discusses emerging reforms, technological advancements, and policy initiatives that may strengthen the Indian biosimilar ecosystem. Future perspectives emphasize the need for enhanced pharmacovigilance, regulatory convergence, streamlined approval pathways, and increased stakeholder confidence to position India as a global leader in biosimilar innovation and manufacturing. Overall, a balanced and science-driven regulatory strategy is essential for ensuring patient safety, fostering innovation, and expanding access to cost-effective biologic therapies.
Biological products, also referred to as biologics, have transformed modern therapeutics with highly targeted and effective therapeutics for a large number of chronic and life-threatening diseases. In contrast to the traditionally chemically synthesized small molecule drugs, biologics are complex molecules generated by means of a sophisticated biotechnology process involving the use of living organisms like bacteria, yeast, or mammalian cells. They are monoclonal antibodies, recombinant proteins, vaccines, hormones, blood components and gene-based therapeutics. Biologics have revolutionized the treatment of many conditions, including cancer, rheumatoid arthritis, diabetes, autoimmune disorders, inflammatory bowel disease, psoriasis and rare genetic disorders, because of their high specificity and therapeutic effectiveness. Biologics are one of the fastest-growing sectors of the global pharmaceutical market, and have played a significant role in helping to make patients' lives better in the last 20 years [1]. Biologics have been linked with very high manufacturing costs, high clinical development costs, and complex manufacturing technologies. Consequently, the uptake of these therapies is limited, particularly in developing and low-income countries. Many blockbuster biologics have come to an end of their patent protection and as a result, biosimilars have emerged as biologic products that are very similar to an approved biologic reference product, in terms of quality, safety, and efficacy [2]. The complexity and variability of biological production processes means that biosimilars cannot be identical to the innovator biologics. They are, however, designed to show no clinically meaningful difference to the reference product via extensive analytical, non-clinical and clinical comparability studies [3].
The introduction of a new class of medical products, called biosimilars, has generated a significant and growing global interest as healthcare systems strive to reconcile the growing costs of therapeutic use with the growing demand for more potent and effective biologic therapies. The regulatory authorities, like the European Medicines Agency (EMA), the United States Food and Drug Administration (USFDA) and the World Health Organization (WHO) have set up a dedicated pathway for the approval of biosimilar products to ensure the quality and safety of these products to the patients [4]. The regulatory framework for biosimilars was first established in Europe in 2005 and has since seen the approval of many biosimilars in a variety of therapeutic categories. Likewise, in 2009, the United States enacted the Biologics Price Competition and Innovation Act (BPCIA) to establish a shortened approval process for biosimilars. The global biosimilar market has grown at a rapid pace because of the expiration of patents, the rising healthcare cost, advancement of manufacturing technologies, and a growing acceptance from healthcare professionals and patients [5]. Worldwide, the trend toward the use of biosimilars has translated into substantial economic savings. Product competition and cost savings in treatment with biosimilars contribute to significant healthcare savings and the fact that biosimilars represent cost-effective alternatives to high-priced originator biologics makes them cost-effective. Biosimilars have improved access for patients to biologic treatments which were too costly for many populations in many countries. Moreover, with the emergence of biosimilars, pharmaceutical companies must innovate further to create better therapeutic products and manufacturing processes. This global biosimilar market is projected to see an exponential rise in the future years because of the growing incidence of chronic diseases, growing geriatric population and supportive regulatory initiatives [6]. India is fast becoming one of the major players in the global biosimilar industry, thanks to its advanced pharmaceutical manufacturing facility, skilled human resources, and low manufacturing costs. India's pharmaceutical industry is known to be one of the world's biggest manufacturers of generic drugs and it has helped India move towards biosimilar development and manufacturing. India had already approved one of the world's oldest biosimilars before the formal guidelines were drawn up.India has been one of the first countries to approve a biosimilar before the formal guidelines were introduced. Indian pharmaceutical organizations like Biocon, Dr. Reddy's Laboratories, Intas Pharmaceuticals, Zydus Lifesciences and Reliance Life Sciences have been playing a significant role in the development and commercialization of biosimilars in the country as well as internationally [7]. Biosimilars have a special place in the Indian healthcare scenario due to the population, growing burden of chronic diseases, and socio-economic inequality in access to healthcare in India. The prevalence of diseases like cancer, diabetes, rheumatoid arthritis and autoimmune diseases are on the rise in India, resulting in an increasing need for affordable biologic therapies. Yet, because of the expense of innovator biologics, many patients can't afford these treatments. Biosimilars offer a viable option in lowering treatment expenses while remaining to be equally effective and safe. Lower cost biosimilars can make healthcare more accessible, cut out-of-pocket costs and help government healthcare programs deliver universal health coverage [8]. To tackle the rising tide of the development of biosimilars, the Central Drugs Standard Control Organization (CDSCO), under the Department of Biotechnology (DBT) in India has developed an exclusive regulatory body for biosimilars. In 2012 and 2016, the “Guidelines on Similar Biologics” were issued to establish a scientific and regulatory course of action for demonstrating biosimilarity. The guidelines describe requirements for quality characterization, manufacturing processes, non-clinical evaluation, clinical studies, post-marketing surveillance. The Indian regulatory system is designed to ensure that biosimilars are of acceptable quality, safety and efficacy prior to approval and commercialization [9]. India has come a long way in developing the biosimilar regulations, but there are still challenges and gaps. Despite progress, a number of concerns remain, including the strengths and weaknesses of pharmacovigilance systems, policies on interchangeability, its implementation by regulators, transparency in regulatory processes and limited harmonisation with international standards, among others, that continue to worry healthcare professionals and experts. Furthermore, the complexity of the manufacturing process for biologic drugs and the possibility of immunogenicity necessitates rigorous quality control and post marketing surveillance to assure patient safety [10]. Given the dynamic growth of the biosimilar market and India's emerging position in the international pharmaceutical market, an urgent need for a critical assessment of the current regulatory landscape for biosimilar approval in India is felt. To enhance healthcare access, foster innovation and maintain public trust in biosimilar products, it is crucial to understand the strengths and weaknesses of the Indian biosimilar regulatory landscape, along with future prospects. Hence, the aims of this review are to give a comprehensive overview of the regulatory framework for biosimilar approval in India, highlight any gaps and challenges, and explore future prospects for enhancing the biosimilar regulatory landscape to keep pace with the changing global landscape [11].
Concept and Characteristics of Biosimilars
Biologic products developed to be highly similar to an already approved reference biologic product in terms of quality, safety, efficacy and biological activity are known as biosimilars or “similar biologics” in India. Biologics are produced in a high tech biotechnological process involving living cells, typically bacteria, yeast or mammalian cells, unlike conventional pharmaceuticals which are synthesized chemically. Biosimilars are launched after patent off the innovator biologics, with similar clinical properties as innovators, and with lower costs. The regulatory authorities, including the World Health Organization (WHO), the European Medicines Agency (EMA), and the United States Food and Drug Administration (USFDA), give the following definition for a biosimilar drug: It is a drug that is identical to the reference drug with respect to its purity, potency, and safety [12]. Biologics, biosimilars, and generic drugs have a significant difference. Generic medicines are chemically identical to the small molecule and have the same active ingredient, dosage form and pharmacological properties as the original product. Small-molecule drugs have simple, well-defined chemical structures, which makes it easy to replicate them exactly by chemical synthesis and thus produce generic versions. Biologics are much more complex molecules that have large molecular weight and complex 3D structures, and are synthesized in a living system. This complexity and variability of living systems make it impossible to have biosimilars that are identical to the innovator biologic [13]. Rather, they are intended to be quite comparable, and there are no important differences in clinical effectiveness or safety. One of the most significant features of biologic products that affects the development and regulation of biosimilar products is the complexity. Biologics may be proteins, glycoproteins, monoclonal antibodies, or products derived from recombinant DNA which have to have complicated post-translational modifications (glycosylation, folding, and phosphorylation). The biological activity, stability, or safety profile of the product can vary from its last product depending on minor changes in cell lines, fermentation conditions, purification procedures, or storage conditions [14]. As a result, manufacturers must use state-of-the-art analytical characterization methods and strict quality control procedures to guarantee consistency between the biosimilar and the reference biologic. Multiple comparability exercises are required by regulatory authorities for physicochemical, biological and functional testing to ensure biosimilarity. Another important issue related to biologics and biosimilars is immunogenicity, meaning the ability of a therapeutic product to stimulate an immune response in the recipient [15]. Biologics may trigger anti-drug antibodies, which can lower the efficacy of the drug and/or lead to side effects because they are protein-based products. The immunogenic responses may depend on structural changes, impurities, formulation changes, storage conditions, route of administration, and patient-related variables. In certain cases, immunogenicity can have serious clinical implications such as hypersensitivity reactions or immune-mediated blocking of endogenous proteins. Thus, immunogenicity is an essential aspect of biosimilar development and assessment for regulatory purposes. In order to reduce immunogenic risks, regulatory authorities have asked for comparative immunogenicity studies that are conducted throughout the clinical development process and ongoing pharmacovigilance following market approval. Manufacturers will need to prove that the biosimilar does not have a higher immunologic risk than the reference product. There is also a need for post-marketing surveillance systems in order to identify rare or late-occurring immune-related adverse effects. Biosimilars, in general, are a high-tech and cost-saving therapeutic class, which brings the advantages of biologic therapy along with increased cost-effectiveness and accessibility. They are very complex, however, and may be immunogenic, which makes them very sensitive to proper regulation for quality, safety and therapeutic equivalence [16-20].
Evolution of Biosimilar regulation in India
The regulatory landscape for biosimilars in India is evolving, and the country's capabilities in the field of biotechnology, pharmaceutical production, and healthcare innovation are expanding. India was one of the first nations to see the therapeutic and economic promise of biosimilars, especially in the face of the growing burden of chronic diseases and the prohibitive cost of innovator biologic therapies. The Indian regulatory system has undergone extensive changes and transformation over the years to create a scientifically sound regulatory framework for the quality, safety and effectiveness of biosimilar products [21]. In the early regulatory landscape, India had regulation of biosimilars under the general provisions of the Drugs and Cosmetics Act, 1940 and Drugs and Cosmetics Rules, 1945. There were at the time no biosimilar-specific guidelines and evaluation of biologic products was done in a similar manner to conventional drugs. There was a lack of specific regulations which led to uncertainties about approval pathways, quality standards, clinical evaluation requirements and pharmacovigilance practices. However, in spite of all these restrictions, India has approved some recombinant biologic products, such as vaccines, insulin, erythropoietin, and growth hormones, before the many other developing nations. Lower manufacturing costs, availability of skilled workforce as well as increasing domestic demand for cheaper biologic drugs contributed to the rapid growth of the biotechnology capabilities of Indian pharmaceutical companies. It was noticed that a comprehensive regulatory framework for biosimilars is needed, which was established in 2012 by the Central Drugs Standard Control Organization (CDSCO) and the Department of Biotechnology (DBT) in collaboration. These guidelines marked a significant step in the evolution of Indian biosimilar guidelines and were largely in line with the recommendations of the World Health Organization (WHO). The guidelines of 2012 set a stepwise approach for approval of biosimilars, which involves detailed analytical characterization, preclinical testing, clinical trials and post-marketing surveillance. The framework prioritized showing similarity of an existing, approved reference biologic, over proving safety and efficacy from scratch. This enabled a scientific and patient-safe approach for a lower development cost. The Indian Biosimilar guidelines were updated in 2016 to reflect the advances in the science of biosimilars and regulatory issues. The updated guidelines brought more clarity into the reference biologic selection, analytical similarity and pharmacovigilance requirements. A key change was the ability to sub-opt for some of the animal toxicity tests if there was sufficient scientific justification and analytical comparability data. The 2016 revisions also bolstered the post-marketing surveillance obligations, and promoted the adoption of advanced analytical technologies for further characterization of biosimilars. In addition, flexibility was added in the confirmatory clinical trial requirements that depend on the extent of evidence produced in the course of the comparability assessment. The changes were intended to reduce the development timelines for biosimilar products, without compromising on the safety and quality of the products for patients. In recent times, the draft revisions for 2025 have been discussed, suggesting a more sophisticated and standardized approach to regulation for India. The proposed changes focus on the need for risk-based regulation, analytical characterization, minimization of unnecessary animal use and the greater use of real world data. Regulatory authorities are also increasingly looking at digitalisation of regulatory submissions, modernisation of pharmacovigilance systems and compliance with international standards set by EMA, USFDA and WHO [22]. The use of sophisticated manufacturing technologies and Quality by Design (QbD) is likely to further reinforce the biosimilar regulations in India. In the last decade, the Indian biosimilar market has grown significantly, and it is emerging as one of the biggest biosimilar market in the world. Biosimilars are being developed by Indian pharmaceutical companies like Biocon, Dr. Reddy's Laboratories, Intas Pharmaceuticals, Zydus Lifesciences and Reliance Life Sciences for oncology, diabetes, autoimmune disorders and hematological diseases. The cost of the medicines and robust manufacturing facilities have helped India emerge as a major exporter of biosimilars to international markets. The growing biosimilar industry not only enhances patients' access to cutting-edge biologic drugs but also plays a pivotal role in driving the growth of the Indian pharmaceutical and biotechnology sector [23].
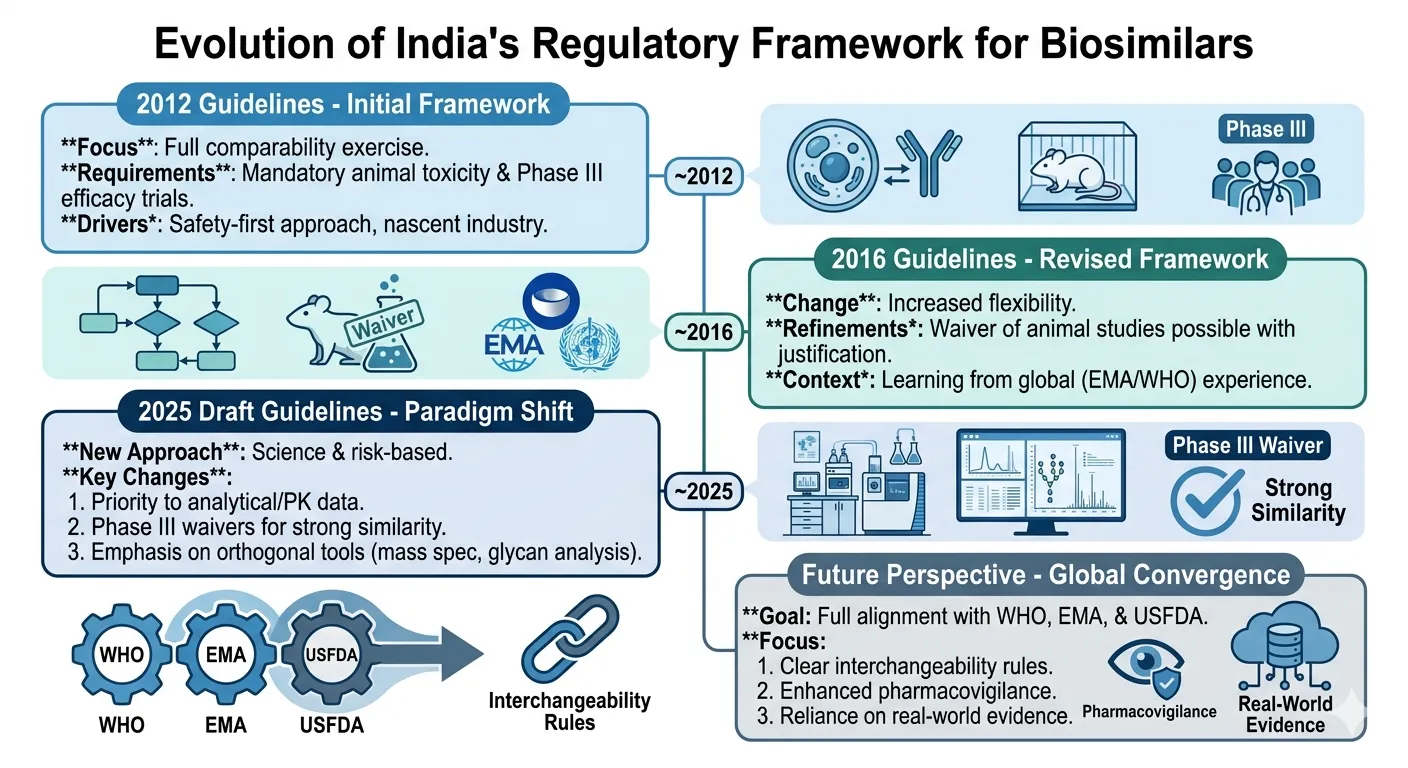
Fig.1: Evolution of India's Biosimilar Regulatory Framework
Regulatory Authorities Governing Biosimilars in India
Biosimilars regulation in India is a multiagency and multiscientific approach involving several Governmental bodies and scientific committees to ensure the quality, safety and efficacy of bio-products and ethical compliance. The approval and monitoring of biosimilars is complex and has specific scientific requirements and regulatory control, since they are extremely complex therapeutic products of a biological origin [24]. The important regulatory bodies for biosimilar Governance in India are Central Drugs Standard Control Organization (CDSCO), Department of Biotechnology (DBT), Review Committee on Genetic Manipulation (RCGM), Drug Controller General of India (DCGI), and Institutional Ethics Committees (IECs). India's main regulatory authority for the control of pharmaceuticals and biologic products is the Central Drugs Standard Control Organization (CDSCO). CDSCO is the regulatory authority which is responsible for approving new drugs, clinical trials, manufacturing license, import permission and post marketing surveillance activities under the Ministry of Health and Family Welfare. For biosimilars, CDSCO reviews the quality, safety and efficacy information provided by the manufacturer using the “Guidelines on Similar Biologics.” Prior to market authorisation, the organisation ensures that Good Manufacturing Practices (GMP), pharmacovigilance and regulations are complied with. The CDSCO also keeps an eye on adverse drug reactions and co-ordinates post marketing safety surveillance to ensure the safety of patients in the future. The Department of Biotechnology (DBT), under the Ministry of Science and Technology, is one of the significant regulatory departments for biotechnology derived products. DBT helps deliver scientific and technical guidance on recombinant DNA technology, genetic engineering and biologic manufacturing processes. The department works closely with CDSCO with respect to the preparation of guidelines and revision of the guidelines for biosimilar. Additionally, DBT promotes biotechnology research and innovation while ensuring that biologic development complies with biosafety and ethical standards. DBT's science enables the growth of India's biotechnology and biosimilar industry. One of the most significant scientific bodies that work under DBT is the Review Committee on Genetic Manipulation (RCGM). RCGM is responsible for the surveillance of research on genetically engineered organisms (GEOs) and recombinant DNA technologies (RDTs). RCGM review and approval is needed for biosimilars made by recombinant methods at the early preclinical and development stages. The committee considers biosafety issues, environmental risks, laboratory containment measures, and adherence to the norms of genetic engineering. RCGM monitors biotechnology research operation, for the safe development and handling of genetically modified biological products. The Drug Controller General of India (DCGI) is the top-level authority of CDSCO and is the central licensing authority of new drug or biosimilar in India. The DCGI has a pivotal role in granting approval to clinical trials, manufacturing licenses, import and final market authorization of biosimilar products. As part of the process of evaluation, the DCGI examines scientific information on analytical characterization, non-clinical studies, clinical trials and pharmacovigilance plans. Institutional Ethics Committees (IECs) have a pivotal role in ensuring the ethical adherence to clinical research with biosimilars. Hospitals, medical institutions and clinical trial centers have these committees which will review clinical trial protocols before the studies start. IECs evaluate informed consent procedures, patient safety measures, risk-benefit analysis, confidentiality protections and compliance with Good Clinical Practice (GCP) guidelines. They are responsible for the protection of participants' rights, dignity and welfare during the conduct of trials. These regulatory authorities form a complex and multi-layered regulatory framework for biosimilars in India. They work in a coordinated manner, providing scientific rigour, biosafety, ethical compliance and ongoing monitoring during the biosimilar products lifecycle [25].
Legal and regulation framework
The legal and regulatory guidelines for the manufacture of biosimilars in India are drawn up to guarantee the quality, safety, efficacy and ethical evolution of biologic products. Because biosimilars are complex therapeutic products manufactured by biotechnology processes, a blend of pharmaceutical laws, clinical trial regulations, manufacturing standards and provisions of intellectual property are necessary to regulate their production. Indian regulatory framework is largely governed by the Drugs and Cosmetics Act, 1940, Drugs and Cosmetics Rules, 1945, New Drugs and Clinical Trials Rules (NDCTR) 2019 and the “Guidelines on Similar Biologics” released by the Central Drugs Standard Control Organization (CDSCO) and the Department of Biotechnology (DBT) [26]. The Drugs and Cosmetics Act, 1940 is the basic act passed by the Government of India for the regulation of drugs, cosmetics and biologic products. The Act gives the legal power to control the import, manufacturing, distribution and sale of pharmaceutical products for the safety of the public and quality of the products. Biological products include biosimilars and are subject to the provisions of this Act. The bill provides powers to the regulatory bodies like CDSCO and the DCGI for evaluation and approval of biosimilar drugs before putting them into the market. The Drugs and Cosmetics Rules, 1945 were framed under the Act which contained detailed procedural and technical conditions for drug approval, manufacturing, drug labelling, drug testing, storage and clinical trials. The rules establish standards for licensing procedures, quality control and manufacturing, for biologics and biosimilars. One of the major developments in the Indian regulatory regime was the enactment of the New Drugs and Clinical Trials Rules (NDCTR) 2019. The rules were created to speed up and streamline the approval process for new drugs, including those of a biological or biosimilar nature. NDCTR 2019 clarifies the requirements for clinical trial permissions, ethics committee registration, compensation for injuries during a clinical trial, accelerated approval modalities, and access to investigational products after the trial. The rules are designed to: improve transparency, increase the efficiency of regulation, and promote innovation, without compromising patient safety. The Drugs and Cosmetics Rules contain the guidelines and requirements for conducting clinical trials in India (Schedule Y) [27]. It outlines comprehensive standards on Good Clinical Practice (GCP), informed consent process, responsibilities of ethics committees, reporting adverse events and documentation of clinical studies. Biosimilars manufacturers have to follow Schedule Y requirement during comparative clinical trials and immunogenicity studies. The guidelines provide for scientific and ethical validity and monitoring of clinical trials conducted with biosimilars. Another important part of biosimilar regulatory considerations is Good Manufacturing Practices (GMP) compliance. Biologics are very sensitive to process, so ensuring product quality is critical. Indian pharmaceutical manufacturers of biosimilars will have to meet the GMP standards laid down in Schedule M of the Drugs and Cosmetics Rules. The GMP standards are applicable to the design of the facilities, validation of equipment, quality assurance systems, personnel training, environmental controls and documentation procedures. Strict adherence to GMP will help reduce the chance of contamination, variability and inconsistencies in manufacturing. Another aspect of biosimilar regulation is intellectual property and patent issues. There is a general rule that biosimilars are copied after the expiration of the patents that protect innovator biologics. Biologics, however, typically have complicated patent environments in terms of manufacturing process, formulation and therapeutic application. India has adopted the Patent Act, 1970, that strikes an appropriate balance between protection of innovations and public health. India currently doesn't offer data exclusivity for biologics, which can help speed biosimilar entry to the market earlier as compared to some developed countries. In conclusion, the Indian legal and regulatory landscape for biosimilars is designed to strike a balance between ensuring affordability, fostering innovation, patient safety, and scientific integrity, while also encouraging the development of the biotechnology and pharmaceutical industry [28].
Similar Biologics in India
The key guidelines for approval of biosimilars in India are the “Guidelines on Similar Biologics” issued jointly by the Central Drugs Standard Control Organization (CDSCO) and the Department of Biotechnology (DBT). These guidelines were first issued in 2012 and updated in 2016, and provide scientific principles to establish biosimilarity to an approved reference biologic product. The framework is based on the recommendations of the World Health Organisation (WHO) and is based on a step-wise comparability approach consisting of quality, non-clinical and clinical evaluations. One of the fundamental ideas in the guidelines is the notion of a reference biologic product (RBP). The reference product is an existing approved biological product that has quality, safety, and efficacy data.The reference product is a licensed biological product that has a known quality, safety, and efficacy profile which is used as the comparator for the biosimilar. The biosimilar manufacturer will have to show that the proposed product is highly similar to the RBP, in terms of physicochemical properties, biological activity, clinical performance, and immunogenicity [29]. Choosing an appropriate reference product is crucial because all comparability evaluations are made with reference to this product. Comparability exercise is the foundation of biosimilar evaluation and is the subject of guidelines highlighted. The goal of a comparability study is to demonstrate that the biosimilar is not clinically different from the reference biologic. This process is based on the concept of a “totality of evidence” and involves the comprehensive evaluation of data collected from analytical, preclinical and clinical studies. The level of studies that need to be done will vary depending on how much similarity was shown at each level. If there is a strong analytical similarity data, it can minimize the extensive clinical studies. One of the most important part of biosimilar development is quality characterization requirements. Advanced analytical techniques need to be used to comprehensively characterize the structural and functional characteristics of the biosimilar. Molecular structure, amino acid sequence, glycosylation pattern, protein folding, charge variants, purity, potency and biological activity are carefully evaluated. The guidelines call for thorough comparison of the biosimilar and the reference product to ensure quality and therapeutic activity. Analytical similarity studies serve an important role in establishment of biosimilarity at the molecular level. The use of sophisticated analytical techniques like mass spectrometry, chromatography, electrophoresis, spectroscopy and bioassays allows for the detection of similarities in structure and function. These studies are useful in determining slight differences that could impact safety or effectiveness. Comparative analytical assessment is regarded as the most sensitive step in biosimilar evaluation as any small difference in the biologic products could affect the clinical outcomes. The biosimilar guidelines also require stability studies. The manufacturer is required to test the stability of the biosimilar under various environmental conditions such as temperature, humidity, light exposure and storage time. Stability studies can be used to evaluate shelf-life, storage conditions and packaging requirements, and can help ensure that the product quality, potency and safety are retained over the product's service life. Biosimilars manufacturing process requirements are very strict because of the complexity of biologic products. Because biologics are made from living cells, minor differences in cell culture conditions, fermentation processes, purification methods or formulation techniques can make a difference in product characteristics. Thus, manufacturers need to create solid and controlled production systems. The regulatory authorities should be provided with detailed information about raw materials, cell banks, process control, purification procedure and contamination prevention measures. Process validation and consistency are important in ensuring batch to batch consistency of biosimilars. Manufacturers need to validate manufacturing processes to ensure consistency of the product over time. Validation is a process of critical process parameters monitoring, in process controls and final product specification monitoring. To reduce contamination risks and to ensure production reliability, Good Manufacturing Practices (GMP) compliance is required. In general, the guidelines for biologic products in India are meant to ensure that biosimilars are scientifically sound and enable the creation of low-cost biologic drugs for widespread patient access [30-32].
Preclinical Evaluation Requirements
The preclinical evaluation is a critical phase of biosimilar development, and it is a crucial step in establishing similarity between a proposed biosimilar, and its reference biologic product. The main goals of preclinical studies are to assess the structural, functional, pharmacological and toxicological properties of the biosimilar before then moving on to human clinical trials. In vitro characterization studies is one of the most crucial aspects of preclinical studies as per ‘Guidelines on Similar Biologics' issued by the Central Drugs Standard Control Organization (CDSCO) in India and Department of Biotechnology (DBT). These studies include both analytical and biological comparisons in detail between the biosimilar and the reference biologic product. Physicochemical characterization is performed by advanced laboratory techniques on molecular structure, amino acid sequence, protein folding, glycosylation pattern, charge variants, purity, and biological activity. Functional assays are also conducted for receptor binding and enzyme activity as well as mechanism of action. The in vitro aspect is considered to be very sensitive to detect the subtle difference between products and is used as a basis for the biosimilar comparability assessment. Animal toxicity studies are performed to assess the safety profile of the biosimilar and to determine if there are any toxicity concerns before exposure of humans. These studies typically consist of repeated dose toxicity studies, local tolerability and an assessment of potential adverse effects on major organ systems. Immunogenicity and reproductive toxicity studies may also be necessary as appropriate for the biologic product. Appropriate animal species selection is essential as biologics may have species-specific activity. In most instances there is only one species of animal in which the toxicity needs to be assessed. The main goal is not to reestablish safety by itself, but to compare the biosimilar to the reference product and determine if there are any unexpected differences [33]. Another subset of preclinical studies is pharmacodynamic (PD) and pharmacokinetic (PK) studies. Pharmacodynamic studies focus on the biological effect and mechanism of action of the biosimilar, and pharmacokinetic studies focus on absorption, distribution, metabolism and elimination properties. Comparative PK/PD studies can help identify if a biosimilar has the same biological activity as the reference product. Where appropriate and scientifically justified these studies may be conducted in animal models. The data from the PK/PD studies assist in the selection of dose and in the planning of clinical trials. Comparative non-clinical evaluation is performed according to the principle that biosimilars should be highly similar to the reference biologic in a head-to-head comparison. All the analytical, functional, toxicological and pharmacological data are considered together by regulatory authorities to decide if there are meaningful differences between the products. The amount of non-clinical testing will vary with the complexity of the biologic and the level of similarity determined in analytical studies. The ethical issues of animal testing have gained greater attention in the context of biosimilar regulation. Biologics are complicated products, and while they might not always reflect human reactions based on conventional animal models. The ethical concerns about animal welfare are also raised by unnecessary and excessive animal testing. In the last years, regulatory initiatives have focused on the 3Rs (Reduction, Refinement, and Replacement) of animal use in research. The new Indian guidelines for biosimilar guidelines suggest that animal studies be minimized, where and when scientifically justified, supported by strong analytical and in vitro data. New cell-based assays, computational modeling and analytical methods continue to help reduce animal use in the development of biosimilars. In conclusion, preclinical evaluation is a critical step in the safety, quality and comparability of biosimilars prior to clinical testing and regulatory approval [34].
Clinical Evaluation of Biosimilars
Clinical evaluation is an essential part of the biosimilar development process that seeks to establish that the clinical safety, efficacy and immunogenicity of the proposed biosimilar product are not clinically different from the reference biologic product. The process of biosimilar evaluation is more similar to the process of drug evaluation in which much less independent clinical evidence is needed, except that it is based on a “totality of evidence” approach that takes into account the extent of the clinical experience. The clinical evaluation is guided by the “Guidelines on Similar Biologics” by the Central Drugs Standard Control Organization (CDSCO) and the Department of Biotechnology (DBT) and in most of the countries Phase I clinical studies are the first human studies performed during the clinical evaluation of biosimilar products [35]. These studies are mainly designed to be the comparison of the pharmacokinetic (PK) and pharmacodynamic (PD) characteristics of the biosimilar with the reference biologic. The objective of a phase I study is to assess safety and tolerability in healthy volunteers or, if required, in patients based on the safety and mechanism of action of the biologic product. Comparative PK/PD studies are a key component of clinical biosimilar evaluation. Parameters that are measured by pharmacokinetic studies include absorption, distribution, metabolism, elimination, maximum plasma concentration (Cmax), and area under the curve (AUC). Pharmacodynamic studies assess the biological activity and therapeutic response by measuring a specific biomarker or clinical indicator. Because of the high levels of sensitivity, these studies are important in identifying a potential difference between the biosimilar and reference biologic. Phase III confirmatory clinical trials are used to demonstrate comparable efficacy and safety of the biosimilar with the reference product in a sensitive patient population, which is the expectation of regulatory authorities. These are usually randomized, controlled, and have sufficient sample size to rule out clinically relevant differences. The condition selected for clinical endpoints should be one which can detect differences in therapeutic performance in these studies. Phase III studies involve safety evaluation including monitoring adverse effects, infusion reactions and long-term treatment outcomes. The overall goal is to establish bioequivalent performance of the biosimilar with the innovator biologic in a clinical setting. One of the most crucial tests performed during clinical evaluation of biosimilars is immunogenicity assessment. As biologics are protein products, they can trigger an immune response in the patient [36]. Immunogenicity may lead to the generation of anti-drug antibodies that could decrease drug efficacy or trigger adverse reactions like infusion reactions, hypersensitivity and autoimmune reactions. So, comparative immunogenicity studies are essential in the course of the biosimilar development. These studies compare the incidence, nature and clinical significance of immune responses to the biosimilar and reference product. Post marketing pharmacovigilance is also important in order to look up for rare or delayed immunogenic effects. One key regulatory term used in the approval of biosimilars is extrapolation of indications. In one therapeutic indication, if biosimilarity can be sufficiently established, regulatory authorities can approve the product in other indications without additional clinical trial for each indication in the same therapeutic category. Extrapolation is based on scientific rationale, such as similarity of mechanism of action, receptor interaction, pharmacokinetics, immunogenicity profile and clinical experiences. This helps to minimize the number of unnecessary clinical studies, limit drug development expenses, and shorten the time for patients to receive an affordable biologic therapy. The new Indian biosimilar guidelines have included flexibility when it comes to waiver of confirmatory clinical trials in some cases. Regulatory agencies can waive or shorten the need for large Phase III efficacy trials if there is extensive analytical characterization and comparative analysis of the PK/PD characteristics that strongly suggests biosimilarity [37]. This risk-based approach is in keeping with the changing regulatory landscape around the world and an increasing level of trust in sophisticated analytical technologies. However, such waiver is allowed only after obtaining enough scientific evidence of absence of clinically meaningful difference between the biosimilar and the reference product. In conclusion, clinical evaluation is still important for demonstrating the safety and efficacy and clinical comparability of biosimilars prior to regulatory approval and commercialization.
Approval Pathway for Biosimilars in India
The approval route for biosimilars is intended to ensure that biosimilar products are of similar quality, safety and efficacy as an approved reference biologic product. The main controller of the regulatory framework is the “Guidelines on Similar Biologics” issued by the Central Drugs Standard Control Organization (CDSCO) and the Department of Biotechnology (DBT) [38]. The approval is done by a step-wise comparability process that is based on analytical, preclinical and clinical evidence. The process of regulatory approvals is done in a step-wise manner starting with identifying an appropriate reference biologic product already approved in India or recognized in international markets. Extensive analytical characterization is a critical step in establishing similarity before moving forward to non-clinical and clinical studies, conducted by the biosimilar manufacturer. The data that are provided through each step support biosimilarity, following the principle of “totality of evidence.” The number of additional animal or clinical studies can be minimized if there is strong analytical and functional similarity. One of the most important aspects of the biosimilar approval process is the application and dossier submission. The manufacturing of the drug should come with a comprehensive dossier to CDSCO, which includes product quality, manufacturing process, analytical comparability, non-clinical studies, clinical trial data, and pharmacovigilance plans. The dossier typically is based on the Common Technical Document (CTD) format and contains administrative data, quality records, preclinical study reports, and clinical data. The details of the raw materials, cell lines, manufacturing controls, stability studies and process validation should also be included. The goal of the submission is to prove that the biosimilar is very similar to the reference product and that there are no clinically relevant differences. Review and Evaluation will include scientific assessment of regulatory authorities and expert committees [39]. Covering analytical characterization, safety, efficacy, immunogenicity, and manufacturing quality, CDSCO reviews the data submitted. The Department of Biotechnology (DBT) and the Review Committee on Genetic Manipulation (RCGM) can also be involved in the evaluation of biosafety aspects and issues relating to recombinant DNA technology. Clinical trial protocols, comparability studies and pharmacovigilance plans are studied by expert committees before approval. Regulators will consider if they need more information, clarification and/or further studies during the evaluation process. Another significant aspect of the approval process is licensing for manufacturing and imports. Under the Drugs and Cosmetics Rules, Schedule M, manufacturers of biosimilars need to adhere to Good Manufacturing Practices (GMP). Manufacturing facilities are audited to guarantee that they have proper QC systems, environmental controls, contamination control and batch consistency. If companies are planning to import biosimilars into India, they should apply for import licenses and submit proof of meeting the Indian regulatory requirements. Imported biosimilars are also subject to quality testing and scrutiny before they can go into the market. Once the regulatory authorities are convinced by the provided evidence of biosimilarity, the market authorization process is started. The Drug Controller General of India (DCGI) grants the approval for commercial marketing after the review of all quality, non-clinical and clinical data [40]. Biosimilars have been approved for therapeutic use for specific indications and in some cases, indications can be extrapolated from scientific arguments. Product labelling, prescribing information and risk management plans must meet regulatory requirements prior to commercialisation. Post-marketing monitoring and the requirement to conduct Periodic Safety Update Reports (PSURs) and implement pharmacovigilance programs are part of manufacturer's responsibilities. Monitoring helps to detect unusual or late occurring effects, especially immunogenic effects, if any. In addition, regulatory authorities can carry out post-marketing inspections, and ask for further safety and efficacy data if needed. The overall goal of the Indian biosimilars approval process is to ensure scientific rigor, patient safety, cost-effectiveness, and timely access to biologic drugs while fostering the development of the biotechnology industry [41].
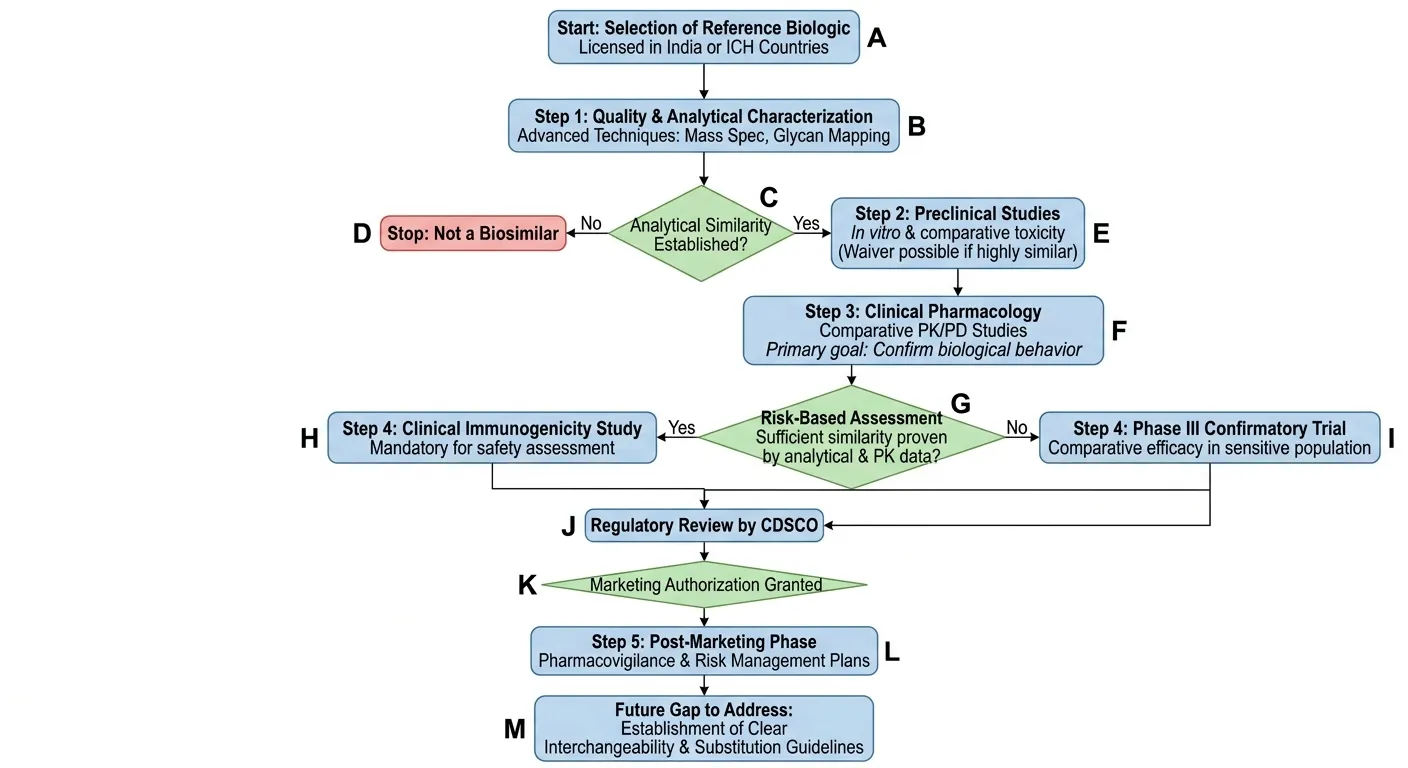
Fig.2: Stepwise approval pathway for biosimilars in India
Pharmacovigilance and Post-Marketing Surveillance
The regulation of biosimilars is critical as they are very complex, and adverse drug effects can be identified after the drug has been marketed. Pharmacovigilance and post-marketing surveillance are key aspects of biosimilar regulation [42]. While biosimilars must be extensively analyzed, tested, and studied in preclinical and clinical trials before approval, they still need to be monitored after approval to demonstrate their continued safety and efficacy in real world clinical use, as well as to ensure quality. Risk management plans (RMPs) are an important aspect of the post marketing safety surveillance of biosimilars. Comprehensive risk management strategies need to be developed by manufacturers to identify potential safety concerns that might arise with the biosimilar and how to minimize the risks. These plans involve the implementation of safety monitoring systems, adverse event reporting systems, patient education programs, and risk management strategies for identified or perceived risks. Biologics are characterized by the fact that small manufacturing differences could have an effect on the immunogenicity or therapeutic effect and therefore periodic risk management plans (PMPs) are required regulatory documents to be submitted by biosimilar manufacturers following market authorization (PAMPs). PSURs are meant to give more up-to-date details about how the biosimilar has been used after it was first made available. These reports cover information on adverse drug reactions, serious adverse events, medication errors, drug ineffectiveness and new safety signals. Biosimilar manufacturers are normally expected to design PSURs for every six months for two years after approval and after that every year for a given period based on regulatory requirements in India. One of the critical components of the pharmacovigilance of biosimilars is adverse drug reaction (ADR) monitoring. Adverse events related to biologic therapies are encouraged to be reported via the following Pharmacovigilance Programs: Healthcare Professionals (HWP) and Manufacturers (MPs) can report adverse events through the Pharmacovigilance Programme of India (PvPI) and Patients (PPI) can report through the Adverse Drug Reactions Programme of India (ADRPI). Biosimilars can lead to immunological reactions, allergic reactions, infusion-related reactions, or unanticipated side effects that need to be monitored continuously [43-45]. Traceability and naming are significant problems in the field of pharmacovigilance with regard to biosimilars. There can be more than one biosimilar to the same reference biologic, so it is important to have the correct identification of the specific product to which an adverse event is related. Traceability is the ability to keep a careful track of product name, manufacturer, batch number and prescribing information to allow for proper monitoring of adverse reactions. Lack of traceability may make it difficult to assessment causality and may also be a barrier to the effective implementation of pharmacovigilance activities. The generation of real world evidence has become a key aspect in the evaluation of biosimilars long term safety and efficacy. Real world evidence is obtained from clinical practice data, patient registers, EHR, observational studies and healthcare databases. This evidence yields important clues about treatment response, switching, immunogenicity, and long-term safety in a variety of patient populations [46]. The importance of the use of real-world evidence for post-marketing surveillance and regulatory decision-making is increasingly understood by regulatory authorities. Even with the improvements made there is still a need for improvement in pharmacovigilance infrastructure for biosimilars in India, such as the underreporting of adverse events, lack of awareness among healthcare professionals, problems with traceability etc [47]. In order to set up a proper post-marketing surveillance, it is important to strengthen pharmacovigilance programmes, optimise digital reporting systems, improve training of healthcare professionals and increase patient awareness. Overall, pharmacovigilance and post-marketing surveillance are essential to provide the confidence of the public in biosimilars and to guarantee their safe and effective application in clinical practice [48-50].
Challenges and Regulatory Gaps in India
Even though there have been substantial progress towards development of a regulatory framework for biosimilars, there are several challenges and regulatory gaps that may impact the quality, safety and international acceptance of biosimilar products in India. With the boom of the biosimilar field, these restrictions are crucial for maintaining patient trust, regulatory acceptance, and market competitiveness [51]. The biggest difficulty is the lack of harmonization from various regulatory standards set by the World Health Organization (WHO), European Medicines Agency (EMA), and United States Food and Drug Administration (USFDA). The Indian “Guidelines on Similar Biologics” are overall similar to the global recommendations, but there are certain differences with regard to clinical trials, interchangeability, pharmacovigilance, and analytical comparability. These discrepancies can hinder Indian manufacturers to get global market approvals and can also impact the international confidence in the Indian biosimilars. Another crucial issue in the Indian biosimilar framework is the variability in interpretation of regulations. Biosimilars are very complex scientific products and the regulatory decisions often need to be case by case. The interpretation of individual expert committees, reviewers and regulatory authorities may cause inconsistencies in approval procedures and requirements. Sometimes, managers will be unsure of what analytical, preclinical and/or clinical evidence is needed to approve the product [52-55]. Lack of pharmacovigilance infrastructure is one of the biggest issues in India. While over time the post-marketing surveillance system has also improved, underreporting of adverse drug reaction is still a major issue. Healthcare professionals and patients are not very aware of biosimilar pharmacovigilance and reporting of adverse events procedure. The long-term monitoring of biosimilars is also hindered by inadequate digital reporting systems, poor traceability systems and a lack of real-world safety data. Enhancing the pharmacovigilance infrastructure is crucial to uncover the rare or delayed adverse effect, especially immunogenic adverse effects. One of the other hurdles is the lack of transparency in the approval process for biosimilars [56]. The information available to the public on the process of regulatory review, clinical evaluation data and scientific basis for approvals is often limited. Assessment reports and scientific review documents are not necessarily available in India like regulatory bodies like EMA or USFDA. Low transparency could result in stakeholder distrust and concerns over the consistency and quality of decision making. Another regulatory gap is those concerning interchangeability and substitution. Currently, there are no clear and comprehensive guidelines for automatic substitution of biosimilars at the pharmacy level in India. Interchangeability means that you can change from a biosimilar to its reference product or vice versa without affecting its safety and effectiveness. Limited knowledge of and uncertainties regarding specific interchangeability standards may result in uncertainty among healthcare professionals and reduced uptake of biosimilars by physicians and patients. Biologics are very sensitive to manufacturing conditions, and manufacturing and quality consistency concerns are important. The structure, biological activity and immunogenicity of the product may differ slightly from one cell line to another, depending on differences in the purification procedure or the production process [57]. The development of sophisticated technology, the use of highly controlled manufacturing environments and tight application of Good Manufacturing Practices (GMP) are essential to ensure batch-to-batch consistency. New guidelines for biosimilars, which require fewer clinical data, have also raised questions among smaller manufacturers. While fewer clinical trials could speed the process of approval and ease development costs, some experts have concerns about lack of long-term safety and efficacy data. Rare clinical or immunogenic effects in different patient populations may not be adequately captured by relying on analytical comparability exclusively. Another challenge is the ethical and legal issues raised with the regulation of biosimilars in India [58]. Problems of informed consent, patient safety during clinical trials, patent protection, data protection and biosimilar substitution policies still need to be carefully monitored. Affordability, innovation and the protection of patients is a big challenge facing policy makers. In summary, filling these regulatory gaps to ensure more effective harmonisation, transparency, better pharmacovigilance management, and a more comprehensive policy guidance will be key to the long-term development of the Indian biosimilar market [59, 60].
Future Perspectives
The regulatory landscape of biosimilars in India is intrinsically tied to its capability in improving its regulatory system, global best practices and scientific invention. India has immense potential to become a center of excellence for the development and manufacturing as well as export of biosimilars, given the growing global demand for affordable biologic therapies. But for this to happen, it is essential that regulations are continually updated, pharmacovigilance systems are robust, cooperation is strengthened across countries, and the confidence of stakeholders is raised. One of the most important priorities in the future is harmonisation with the international standards set by the World Health Organization (WHO), the European Medicines Agency (EMA) and the United States Food and Drug Administration (USFDA). Indian Biosimilars would be internationally accepted and credible if they met global regulatory standards. Analytical comparability, clinical evaluation, pharmacovigilance, interchangeability and manufacturing quality harmonized standards can ease the process of market access and minimize regulatory duplication across the globe. International scientific practices would also give a boost to collaborations and investments in the Indian biotechnology industry by multinational partners. Another critical path forward is to improve pharmacovigilance systems. Biosimilars are potentially immunogenic, complex biologic products, where post-marketing surveillance is an essential process to ensure their long-term safety. India needs to strengthen the adverse drug reaction reporting systems, increase the traceability mechanisms, and create strong digital pharmacovigilance platforms. Raising awareness among health care professionals about the safety monitoring of biosimilars and patient participation in adverse event reporting are also crucial. The further strengthening of pharmacovigilance infrastructure through integration of real world evidence, patient registries and electronic health records can also help. The future of biosimilar landscape is expected to witness greater dependency on regulations and collaboration with the international community. The use of assessments and scientific evaluations carried out by trusted international regulatory agencies is called regulatory reliance. Indian regulators can achieve greater efficiency, minimize duplication of reviews, and expedite product approvals by working together with stakeholders like the EMA, the USFDA and WHO. Another key trend that is expected to emerge in the future is the development of faster biosimilar approval processes in India. In some cases, advances in analytical characterization technologies and the increasing trust in comparability science will decrease the need for large animal and clinical studies. Using risk-based regulatory methods and streamlined approval processes can speed up the development process and reduce costs, without compromising patient safety. Accelerating approvals would benefit patients who are seeking cost-effective biologic options and benefit the rapid expansion of the biosimilar industry. Fostering innovation and investment in biotechnology, particularly for sustaining long-term growth, is critical. Government initiatives such as research and development promotions, public-private partnerships, tax benefits, and infrastructure development can boost India's biosimilar environment. New manufacturing technologies, analytical tools based on artificial intelligence and Quality by Design (QbD) practices can enhance product quality and manufacturing consistency. In addition, improving the management and regulatory clarity of intellectual property rights can encourage more domestic and foreign investments.Moreover, improving patient confidence and physician acceptance is also essential to the successful implementation of biosimilars. There still continues to be a degree of uncertainty about the safety, efficacy and interchangeability of biosimilars for many healthcare professionals and patients. Awareness and trust in biosimilars can be enhanced through educational programs, clear communication with the regulatory authorities, and dissemination of scientific data. India has a number of advantages that make it a potential global hub of biosimilar production, such as low-cost production, scientific manpower, generic pharmaceutical expertise, and the growing infrastructure of biotechnology. As long as regulatory measures continue to be strengthened, scientific research is developed and international cooperation is enhanced, Indian companies can become major players in supplying affordable biosimilars for the world.
CONCLUSION
The introduction of biosimilars has been a significant development in contemporary healthcare, bringing down the cost of biologic treatments and enhancing access to these therapies for patients. With its robust pharmaceutical production capabilities, skilled workforce, and cost-effective production infrastructure, India has emerged as a significant market player in the global biosimilar market. The "Guidelines on Similar Biologics" issued by Central Drugs Standard Control Organization (CDSCO) and Department of Biotechnology (DBT) were a significant step towards establishing a robust regulatory framework for the approval of biosimilars in India. These guidelines have made significant contributions to the quality, safety, efficacy and comparability of biosimilar products via analytical, non-clinical, clinical and post-marketing assessments. Indian biosimilars regulatory landscape has undergone significant changes over the years, with a focus on enhancing the scientific rigor, minimizing excess animal testing, bolstering pharmacovigilance, and fostering innovation. The stepwise comparability approach and greater adoption of state-of-the-art analytical technologies is indicative of the biosimilar ecosystem in India becoming more mature. Moreover, the growing Indian biosimilar market has also enhanced the accessibility of the patients to life saving biologic treatments in oncology, diabetes, autoimmune disorders and hematological diseases. While these advances have been made, there are still some regulatory hurdles and gaps that remain. Partially harmonisation with international standards, lack of well-established standards for Pharmacovigilance, lack of clear interchangeability guidelines, variability in regulatory interpretation and problems of transparency are important obstacles. Further, because of the complexity and immunogenicity of biologic products, ensuring manufacturing consistency and ongoing safety monitoring is critical. Strengthening regulatory systems, improving global harmonization, allowing for better post marketing surveillance and more collaboration with global regulatory authorities will be critical to future Indian biosimilar progress. Biotechnology research investment, digital modernization of regulatory processes, Quality by Design (QbD) and real world evidence generation can help further enhance the biosimilar pathway of development and approval. Education campaigns to raise confidence in biosimilars among both physicians and patients will also be critical to ensure their use in the clinic. In conclusion, the landscape of biosimilar innovation, production, and affordable health care delivery in India is poised for a bright future, marked by ongoing regulatory improvements, scientific advancements, and international collaboration, all aimed at meeting patient needs while prioritizing safety and efficacy.
REFERENCES

Aman Raj*, Kriti Dabral, Regulatory Framework for Approval of Biosimilars in India: Gaps and Future Perspectives, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 5, 7469-7489. https://doi.org/10.5281/zenodo.20420207
 10.5281/zenodo.20420207
10.5281/zenodo.20420207