
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1,3 Advanced Institute of Pharmacy, Palwal, Haryana 121105
2 School of Pharmaceutical Sciences, MVN University, Palwal, Haryana 121105
The growing adoption of additive manufacturing in healthcare has enabled the development of 3D-printed medical devices (3D-PMDs) with enhanced customization, design flexibility, and patient-specific applications. However, these innovations challenge conventional medical device regulatory frameworks, which were primarily designed for standardized, mass-produced products. This study presents a systematic comparative analysis of the regulatory frameworks governing 3D-PMDs across three major systems: the U.S. Food and Drug Administration (FDA), the European Union Medical Device Regulation (EU MDR) framework coordinated by the European Medicines Agency (EMA), and standards issued by the International Organization for Standardization (ISO). A qualitative, document-based research approach grounded in regulatory science was employed. Authoritative regulatory documents, guidance papers, legislative texts, and international standards published between 2014 and 2025 were analyzed using thematic content analysis. Comparative matrices, including a regulatory milestone timeline and a gap matrix, were developed to identify areas of convergence, divergence, and regulatory insufficiency. The key gaps were identified in the oversight of point-of-care manufacturing, digital and software-driven design workflows, and validation of patient-specific devices. While ISO standards provide important technical foundations for harmonization, their voluntary nature limits consistent global implementation. This study offers a lifecycle-based comparative perspective on 3D-PMD regulation and highlights priority areas for regulatory alignment to support safe and scalable innovation.
Three-dimensional (3D) printing, commonly referred to as additive or combinatorial manufacturing, has emerged as a significant technological advancement across diverse sectors, particularly in healthcare.[1] In contrast to traditional subtractive manufacturing techniques, 3D printing constructs objects layer by layer using digital design inputs, offering increased flexibility in design, rapid prototyping, and customization.[2], [3] Within the medical field, these capabilities have enabled the development of patient-specific devices, anatomical models, surgical guides, implants, and prosthetics, collectively termed 3D-printed medical devices (3D-PMDs).[4]
The clinical significance of 3D-printed medical devices (3D-PMDs) arises from their capacity to meet unmet medical needs through patient-specific customization and complex geometries that are challenging to produce using traditional manufacturing techniques.[5] Applications of medical 3D printing span a broad range of clinical specialties, with notable adoption in surgical disciplines such as orthopedics, orthopedic oncology, maxillofacial surgery, and neurosurgery, as well as in non-surgical fields including oncology, prosthetics, orthotics, and dentistry.[6] The distribution of these applications across medical specialties is shown in Figure 1. By facilitating the production of patient-matched implants and procedure-specific surgical tools, additive manufacturing has supported improved surgical planning, reduced operative duration, and better clinical outcomes. These developments reflect a significant shift in how medical devices are designed, manufactured, and implemented in clinical practice.
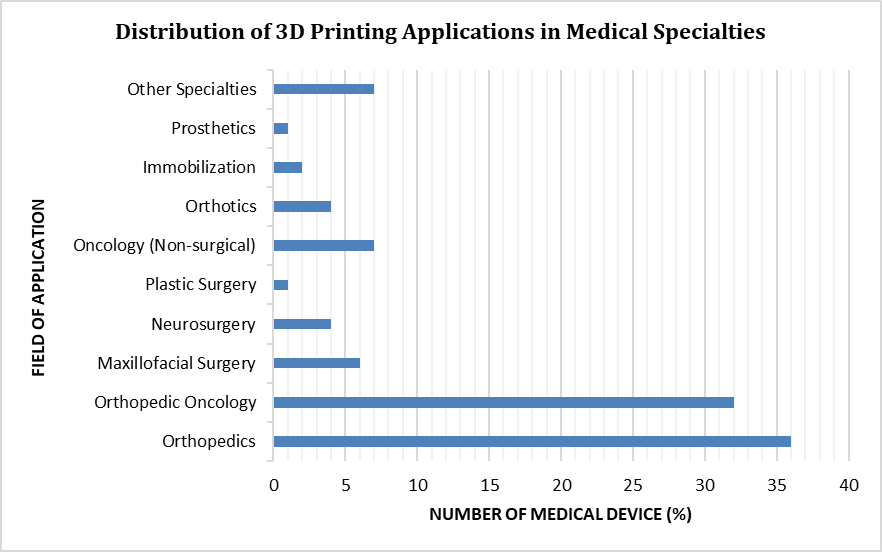
Figure 1. Distribution of 3D Printing Applications in Medical Specialties
Despite these advantages, the regulatory oversight of 3D-PMDs presents substantial challenges.[7] Traditional medical device regulatory frameworks were largely developed around standardized, mass-produced products with fixed designs and controlled manufacturing environments.[8], [9] In contrast, 3D-PMDs are often characterized by customization, variability in design inputs, dependence on digital workflows, and, in some cases, decentralized or point-of-care manufacturing within healthcare institutions.[10] These characteristics complicate established regulatory assumptions related to device classification, manufacturing validation, quality assurance, and post-market surveillance.[11]
A key distinction between conventional medical devices and 3D-printed medical devices (3D-PMDs) lies in their manufacturing paradigm. Traditional medical devices are generally produced in large batches using standardized and well-controlled processes, whereas 3D-PMDs may be manufactured in small batches or as single, patient-specific units derived from medical imaging data and computer-aided design (CAD) models.[12], [13], [14] This shift introduces important regulatory challenges related to reproducibility, consistency, and traceability, particularly when manufacturing parameters, material properties, and post-processing steps vary across devices. Consequently, regulatory authorities are increasingly required to evaluate not only the final product but also the complete digital design and manufacturing workflow underlying 3D-PMD production.[15], [16]
Regulatory authorities worldwide have responded to the growth of additive manufacturing in healthcare by adapting existing frameworks and issuing targeted guidance documents.[17], [18] In the United States, the Food and Drug Administration (FDA) regulates 3D-PMDs under its established medical device pathways, including 510(k), De Novo, and Premarket Approval (PMA),[19] while providing additive manufacturing-specific guidance addressing design, manufacturing, and testing considerations.[20] In the European Union, oversight is governed by the Medical Device Regulation (EU MDR 2017/745),[21] which introduced more stringent requirements for clinical evaluation, post-market surveillance, and custom-made and implantable devices categories highly relevant to 3D-PMDs.[22] Complementing these regulatory systems, the International Organization for Standardization (ISO) develops globally recognized standards related to quality management, risk management, biocompatibility, software lifecycle processes, and additive manufacturing technologies.[23], [24], [25]
Although these regulatory frameworks share the common objective of ensuring the safety and performance of medical devices, they differ considerably in regulatory philosophy, scope, and implementation.[26] The U.S. Food and Drug Administration (FDA) applies a risk-based and guidance-driven approach that allows a degree of regulatory flexibility within established quality system requirements.[27] In contrast, the European Union Medical Device Regulation (EU MDR) is based on legally binding conformity assessment procedures implemented through designated notified bodies. ISO standards, while not regulatory instruments themselves, play an important complementary role by establishing technical benchmarks that support regulatory compliance across regulatory authorities.[28] The coexistence of these distinct regulatory systems contributes to a complex and fragmented regulatory environment for manufacturers and healthcare providers seeking to develop, manufacture, or deploy 3D-printed medical devices across multiple markets.[29], [30]
One of the most pressing regulatory challenges associated with 3D-PMDs is the emergence of point-of-care manufacturing.[31] Hospitals and clinical centers are increasingly adopting in-house 3D printing capabilities to produce patient-specific devices on demand. This trend blurs the traditional boundary between manufacturer and healthcare provider and introduces uncertainty regarding regulatory responsibility, quality management oversight, and post-market obligations. Existing regulatory frameworks acknowledge these developments to varying degrees, but clear, harmonized pathways for decentralized manufacturing remain limited.[32]
In addition to manufacturing considerations, the reliance of 3D-PMDs on digital design files and software tools introduces new regulatory complexities. Digital workflows involving image segmentation, CAD modeling, and increasingly artificial intelligence (AI)-assisted design are integral to additive manufacturing but are not fully addressed by conventional medical device regulations. Ensuring data integrity, software validation, cybersecurity, and traceability across the digital lifecycle of 3D-PMDs represents a growing regulatory priority.[33]
Despite the increasing importance of additive manufacturing in healthcare, there remains a lack of systematic comparative analysis examining how major regulatory systems address the unique challenges posed by 3D-PMDs. Existing literature often focuses on individual regulatory body or specific technical standards, providing limited insight into areas of regulatory convergence, divergence, and gaps across global frameworks. Such comparative understanding is essential for identifying barriers to innovation, reducing regulatory uncertainty, and supporting the development of harmonized approaches that can facilitate safe and effective global adoption of 3D-printed medical technologies.[34]
The present study undertakes a structured comparative evaluation of the regulatory frameworks governing 3D-printed medical devices across three major systems: the U.S. FDA, the EU MDR framework coordinated by the EMA, and relevant ISO standards. The study aims to (i) identify and map key regulatory requirements applicable to 3D-PMDs across these frameworks; (ii) compare approaches to device classification, premarket approval, manufacturing validation, and post-market surveillance; and (iii) identify regulatory gaps and areas where harmonization may be required to support innovation while maintaining patient safety. By providing a comprehensive, lifecycle-based comparative analysis, this research seeks to contribute to regulatory science discourse and inform policymakers, regulators, manufacturers, and healthcare institutions navigating the evolving regulatory landscape of 3D-printed medical devices.
2. METHODOLOGY
2.1 Study Design: This study adopted a qualitative, comparative, document-based research design grounded in regulatory science to evaluate the regulatory frameworks governing 3D-printed medical devices (3D-PMDs) across the U.S. Food and Drug Administration (FDA), the European Union Medical Device Regulation (EU MDR) framework coordinated by the European Medicines Agency (EMA), and the International Organization for Standardization (ISO).
2.2 Data Sources: Authoritative regulatory documents, guidance papers, legislative texts, and international standards published between 2014 and 2025 were systematically reviewed. Sources included FDA guidance documents and relevant provisions of 21 CFR, EU MDR 2017/745 and its annexes, and ISO/ISO-ASTM standards relevant to additive manufacturing, quality management, risk management, software, and post-market surveillance. Only official and publicly available documents were included.
2.3 Analytical Framework: A thematic content analysis was performed, organizing regulatory requirements into four lifecycle-based domains:
2.4 Comparative Analysis: A structured comparison was conducted to identify regulatory convergence, divergence, and gaps. Comparative matrices and summary tables were developed to evaluate how each framework addresses additive manufacturing-specific challenges, including customization, software governance, and point-of-care (PoC) manufacturing.
3. RESULT
In this evolving regulatory landscape, three organizations have played a central role in shaping global expectations for the oversight of 3D-printed medical devices: the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA) operating under the Medical Device Regulation (EU MDR 2017/745), and the International Organization for Standardization (ISO), which develops widely accepted voluntary technical standards. Each of these frameworks reflects a different regulatory approach. The FDA follows a risk-based system supported by guidance documents and the Quality System Regulation, allowing flexibility in how requirements are met. In contrast, the EU MDR relies on mandatory conformity assessment procedures and structured oversight, particularly for custom-made and implantable devices. ISO, while not a regulatory authority, provides a consensus-based set of standards that support quality management, risk control, and technical validation across jurisdictions.[35]
3.1 Regulatory Milestones in 3D-Printed Medical Devices (3D-PMDs)
To contextualize the comparative analysis of regulatory frameworks for 3D-printed medical devices (3D-PMDs), it is important to first understand the timeline of how different authorities have responded to the rapid advancement of additive manufacturing technologies. Each regulatory body FDA, EMA, and ISO has developed its approach at different paces and with varying priorities, ranging from technical guidance to legislative reforms and global standardization. [36] Table 1 presents key regulatory milestones from 2014 to 2025, capturing pivotal guidance documents, standard publications, and policy shifts that have influenced the oversight of 3D-PMDs. This timeline provides critical context for evaluating how these frameworks have converged, diverged, or evolved to address the unique challenges posed by 3D printing in healthcare.
Table 1. Regulatory Timeline in 3D-Printed Medical Devices (3D-PMDs)
|
Year |
Regulatory Body |
Milestone |
Description / Impact |
|
2014 |
FDA |
Initial Public Workshop on Additive Manufacturing |
First formal engagement with stakeholders to discuss benefits, challenges, and safety of AM technologies. |
|
2015 |
ISO/ASTM |
ISO/ASTM 52900 Published |
Issued standard terminology for additive manufacturing, laying the foundation for future technical guidance. |
|
2016 |
FDA |
AM Product Approvals Begin |
FDA begins approving 3D-printed devices (e.g., spine implants, facial prosthetics), demonstrating regulatory adaptability. |
|
2017 |
FDA |
Technical Considerations for Additive Manufactured Devices – Guidance Document |
Key FDA guidance outlining design, manufacturing, and testing considerations for AM devices. |
|
2017 |
EU |
EU MDR 2017/745 Adopted |
Introduces explicit provisions for custom-made and implantable devices, affecting 3D-PMDs significantly. |
|
2018 |
ISO/ASTM |
ISO/ASTM 52921 & 52915 Updated |
Defined file formats and coordinate systems essential for regulatory reproducibility of 3D models. |
|
2019 |
EMA |
MDR Transition Begins |
Medical Device Directive (MDD) replaced by MDR, increasing regulatory rigor for custom and innovative devices. |
|
2020 |
FDA |
COVID-19 AM Response |
FDA fast-tracks approval of 3D-printed PPE and ventilator components under EUA, recognizing flexibility of AM. |
|
2021 |
ISO |
ISO 13485 + ISO 20417 + ISO 10993 Updates |
Expanded quality, labeling, and biocompatibility standards relevant to AM devices. |
|
2022 |
FDA |
Discussion Paper on Point-of-Care 3D Printing |
FDA explores future regulatory pathways for decentralized manufacturing, such as in hospitals. |
|
2023 |
ISO/ASTM |
ISO/ASTM 52920 Published |
Standardized qualification principles for AM machines and processes across medical applications. |
|
2024 |
EMA |
Focus on In-House Manufacturing under MDR |
Initiated working groups to address challenges faced by hospitals using 3D printing for patient-specific care. |
|
2025 |
FDA (Projected) |
Final Guidance on Point-of-Care 3D Printing |
Expected release of formal regulatory pathway for 3D printing at hospitals and clinical sites, covering quality systems, validation, and software control. |
|
2025 |
ISO (Expected) |
ISO/TR on AI-Integrated AM Workflows |
Anticipated publication of a Technical Report addressing risk and validation considerations for AI-driven design tools in 3D-PMDs. |
|
2025 |
EMA (Ongoing) |
Implementation Feedback from MDR on Custom-Made Devices |
EU working groups continue refining MDR interpretations related to in-house and patient-specific device production under Article 5(5), especially affecting orthopaedic and dental 3D-PMDs. |
The comparative analysis identified distinct structural approaches adopted by the FDA, EU MDR (EMA), and ISO in regulating 3D-printed medical devices. Regulatory requirements were mapped across four lifecycle domains: device classification, premarket approval, manufacturing and quality systems, and post-market surveillance. A summary of the comparative findings is presented in Table 2.
Table 2. Summary of Thematic Comparison Analysis
|
Regulatory Parameter |
FDA (USA) |
EMA (EU MDR) |
ISO Standards |
|
Device Classification |
Class I–III, includes guidance on 3D-PMDs |
Class I–III, strict on custom implants |
Supports risk-based approach via ISO 14971 |
|
Manufacturing Validation |
21 CFR 820 + 2017 AM guidance |
GSPR + Notified Body audit |
ISO/ASTM 52920, ISO 13485 |
|
Software Oversight |
SaMD regulation, CAD file trace |
MDR Annex IX/XI for software tools |
ISO 62304, ISO 13485 |
|
Customization & POC |
CDE exists, pilot programs active |
Limited guidance, hospitals may be manufacturers |
ISO 52920 guidance only |
|
Post-Market Surveillance |
UDI, adverse event reports |
PSURs, clinical follow-up mandatory |
ISO/TR 20416, ISO 20417 |
3.2 Overview of Regulatory Framework Coverage
3.2.1 Device Classification and Risk Categorization
All three frameworks apply risk-based classification systems. The FDA classifies devices into Classes I–III and provides additive manufacturing–specific considerations within its guidance documents. The EU MDR similarly categorizes devices into Classes I–III, with additional provisions for implantable and custom-made devices. ISO standards do not establish device classification systems but reference risk management principles applicable across device categories.
3.2.2 Premarket Pathways and Conformity Assessment
FDA premarket pathways applicable to 3D-PMDs include 510(k), De Novo, and PMA submissions, with additive manufacturing considerations embedded within existing regulatory pathways. Under the EU MDR, premarket assessment is conducted through conformity assessment procedures involving notified bodies, particularly for higher-risk and custom-made devices. ISO provides supporting technical standards relevant to premarket documentation but does not define approval pathways.
3.2.3 Manufacturing Validation and Quality Management
Manufacturing oversight requirements differ across the frameworks. The FDA applies Quality System Regulation (21 CFR 820) requirements alongside additive manufacturing guidance documents. The EU MDR requires manufacturers to demonstrate conformity with General Safety and Performance Requirements through audits conducted by notified bodies. ISO standards, including ISO 13485 and ISO/ASTM 52920, outline quality management and process validation requirements applicable to additive manufacturing workflows.[37]
3.2.4 Software Oversight and Digital Design Controls
Software oversight provisions were identified across all three frameworks. The FDA includes software considerations through software as a medical device (SaMD) guidance and digital file traceability requirements. The EU MDR incorporates software within its regulatory scope through dedicated annexes, while ISO standards address software lifecycle processes and file integrity through standards such as ISO 62304 and ISO/ASTM 52915.[38]
3.2.5 Customization and Point-of-Care Manufacturing
Regulatory provisions related to customization and point-of-care manufacturing were identified but varied in scope. FDA documents reference decentralized and point-of-care manufacturing models. The EU MDR includes exemptions for in-house manufacturing under Article 5(5). ISO standards currently do not include specific regulatory provisions for point-of-care manufacturing.[39]
3.2.6 Post-Market Surveillance and Traceability
Post-market surveillance mechanisms are addressed across all frameworks. FDA requirements include Unique Device Identification and adverse event reporting systems.[40] The EU MDR mandates post-market surveillance plans, clinical follow-up, and periodic safety update reports.[41] ISO provides guidance on labeling and post-market monitoring through supporting standards.[42]
Summary of Identified Regulatory Gaps
To systematically identify areas of regulatory incompleteness and misalignment, a gap matrix was developed based on the comparative thematic analysis. The matrix maps the extent to which each regulatory framework addresses critical lifecycle elements of 3D-printed medical devices, highlighting areas that are fully addressed, partially addressed, or not addressed. The resulting gaps are summarized in Table 3. This structured comparison helps identify where harmonization, guidance development, or policy reform is urgently needed to enable safe, scalable, and globally consistent adoption of 3D-printed medical technologies.
Table 3. Gap matrix- Key Regulatory Elements vs. Framework Maturity
|
Regulatory Element |
FDA (USA) |
EMA (EU MDR) |
ISO (Global Standards) |
Identified Gap |
|
Risk Classification of 3D-PMDs |
Detailed guidance & device-specific notes |
General classification, limited to MDR Annexes |
Not addressed directly |
Lack of consistent criteria for custom/patient-specific AM devices |
|
Point-of-Care (PoC) Manufacturing |
Under discussion (2021 paper, 2025 expected guidance) |
Covered under exemptions (Article 5.5) |
Not addressed |
No harmonized model for decentralized/in-hospital 3D printing |
|
Process Validation & Quality Systems |
Covered under QSR and premarket guidance |
Requires conformity via notified bodies |
Covered via ISO 13485, ISO/ASTM 52920 |
Misalignment in process validation expectations across jurisdictions |
|
Post-Processing Requirements (Sterility, etc.) |
Mentioned in general AM guidance |
Implied under MDR Annex I |
Fragmented across multiple documents |
No dedicated, integrated guidance on post-processing specific to 3D-PMDs |
|
Software Oversight (CAD, SaMD) |
Robust SaMD and AM file guidance |
Covered under MDR but limited AM specifics |
Partial (file formats, integrity, not AI tools) |
AI-integrated workflows lack clear validation criteria across all bodies |
|
UDI and Traceability |
Comprehensive system in place |
UDI under MDR but inconsistently implemented |
Not applicable directly |
Inconsistent traceability for custom or one-off 3D-printed devices |
|
Biocompatibility and Material Testing |
Detailed under 21 CFR and ISO references |
Rely on general guidance |
Covered under ISO 10993 |
Some AM-specific material risks (e.g., powder residues) not fully addressed |
|
Cybersecurity in Design/ Production Software |
Discussed under general SaMD framework |
Lacks clear linkage to 3D-PMD workflows |
Not addressed |
No defined controls for secure CAD/AM design and transmission processes |
|
International Harmonization |
Some ISO alignment in guidance documents |
MDR refers to ISO, but not formally aligned |
Intended for harmonization |
Regulatory fragmentation limits global scalability and compliance |
As highlighted in the gap matrix, none of the three regulatory systems-FDA, EMA, or ISO provides a fully comprehensive framework that addresses all critical aspects of 3D-printed medical device (3D-PMD) regulation. These gaps underscore the need for stronger coordination across regulatory bodies and more harmonized regulatory approaches, particularly as 3D printing continues to move toward decentralized and patient-specific applications. The following sections examine the broader implications of these regulatory gaps for manufacturers, healthcare providers, and regulatory authorities, and outline strategic directions for improving global regulatory alignment.
DISCUSSION
The comparative evaluation of regulatory frameworks governing 3D-printed medical devices (3D-PMDs) demonstrates that current oversight mechanisms are largely extensions of conventional medical device regulation rather than purpose-built systems designed for additive manufacturing. While this adaptive strategy has enabled early market access and regulatory acceptance of 3D-PMDs, it also reveals structural limitations when applied to technologies characterized by patient-specific customization, decentralized manufacturing, and digitally driven workflows.
Although all examined frameworks employ risk-based regulatory principles, the findings suggest that risk classification alone does not sufficiently capture the multidimensional nature of 3D-PMDs. Unlike traditionally manufactured devices, the safety and performance of 3D-PMDs are influenced not only by intended clinical use but also by variability in design files, manufacturing parameters, post-processing steps, and software-controlled workflows. This creates regulatory tension, particularly for patient-specific implants and one-off devices, where assumptions of batch consistency and standardized validation are difficult to maintain.
A central regulatory challenge identified in this study is the governance of point-of-care (PoC) manufacturing. The increasing adoption of in-hospital 3D printing blurs the traditional distinction between manufacturer and healthcare provider, raising unresolved questions regarding regulatory responsibility, quality management, and post-market accountability. While existing regulatory frameworks acknowledge PoC manufacturing in principle, none provide a fully articulated model that addresses the unique risks associated with decentralized production environments. This absence of clarity may lead to inconsistent compliance practices and uneven patient protection across jurisdictions.[29]
The analysis also highlights significant gaps in the regulation of software and digital design workflows, which are foundational to additive manufacturing. Current regulatory approaches focus primarily on software that performs medical functions, while upstream software used for image segmentation, CAD modeling, and increasingly AI-assisted design remain insufficiently addressed. As AI-driven tools become more integrated into design optimization and patient-specific modeling, the lack of explicit validation, traceability, and cybersecurity requirements represents a growing regulatory vulnerability that extends beyond traditional medical device paradigms.[43], [44], [45]
ISO standards play a pivotal role in supporting regulatory alignment by providing globally recognized technical frameworks for quality management, risk assessment, and additive manufacturing processes. However, their voluntary and non-binding nature limits consistent enforcement and adoption across jurisdictions. While regulators frequently reference ISO standards, variability in how these standards are interpreted and applied contributes to regulatory fragmentation, particularly for manufacturers seeking multinational market access.
From a harmonization perspective, the findings indicate that meaningful regulatory convergence is more likely to emerge through targeted integration of additive manufacturing–specific provisions within existing regulatory systems rather than through the development of entirely new regulatory regimes.[2] Priority areas for alignment include standardized validation of digital workflows, clearer classification criteria for patient-specific devices, and formal regulatory pathways for decentralized manufacturing. Such integration would enhance predictability for manufacturers while maintaining robust safeguards for patient safety.[30], [46], [47]
Importantly, the regulatory gaps identified in this study have direct implications for innovation, clinical adoption, and patient access. Overly rigid regulatory interpretations may delay the availability of personalized devices, while insufficient oversight could compromise device reliability and erode clinical trust. Achieving an appropriate balance between regulatory control and innovation therefore requires adaptive, lifecycle-based oversight models that evolve alongside technological advancements.
Overall, this study underscores the need for regulatory agility and cross-jurisdictional collaboration in governing 3D-PMDs. Strengthening coordination between regulatory authorities, standards organizations, healthcare institutions, and technology developers will be essential to address emerging challenges such as AI-integrated design and point-of-care manufacturing. By aligning technical standards with enforceable regulatory expectations, global regulatory systems can better support the safe, effective, and equitable integration of 3D-printed medical devices into healthcare practice.
CONCLUSION
This paper has systematically analysed the regulatory frameworks governing 3D-PMDs under three key regimes: the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA) under the MDR, and the International Organization for Standardization (ISO). Through thematic comparisons, it is evident that while all three regulatory systems address the core principles of safety and performance, their implementation and scope diverge significantly particularly in areas such as software integration, point-of-care manufacturing, and patient-specific customization. The FDA’s device-specific and adaptive approach offers greater flexibility, especially for developers within the U.S. ecosystem, but remains under development for decentralized manufacturing. The EMA, under its new MDR, has enhanced clinical evaluation and post-market surveillance but raising concerns around the time and cost of compliance. ISO provides a robust foundation of harmonized technical standards but lacks enforcement authority and consistency in adoption across countries. This study provides a foundational comparative analysis of the regulatory approaches to 3D-printed medical devices, illuminating areas of strength, fragmentation, and emerging risk. It contributes to the growing body of literature calling for regulatory agility in the face of rapid technological change. The successful regulation of 3D-PMDs lies not merely in controlling risk, but in enabling innovation while maintaining patient trust and clinical efficacy.
REFERENCES

Ajay Kumar, Shivali Rahi, Arpana Rana, Regulatory Frameworks for 3D-Printed Medical Devices: A Comparative Analysis of FDA, EMA, and ISO Standards, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 1, 2406-2419. https://doi.org/10.5281/zenodo.18341559
 10.5281/zenodo.18341559
10.5281/zenodo.18341559