
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Bhagwan Mahavir College of Pharmacy, Bharthana Road, Surat, Gujarat 395007
The most serious global public health challenge is cancer; hence strong clinical research must be arranged to generate effective remedies. The article compares the regulatory framework and ethical standards governing oncology clinical trials in two countries, namely the United States and India, focusing on the two primary regulatory authorities, U.S. FDA and CDSCO. The difference in approval processes, documentation, and practice in the monitoring of clinical trials is discussed and analyzed. The review examines informed consent process, compensation policies, and adverse event reporting, which are the key components of ethical oversight. The roles of IRBs in the United States and IECs in India, as well as the application of international standards such as ICH-GCP are discussed. The review elaborates on differences in timelines for approval, compliance requirements, and regulatory transparency. Special emphasis has been given to India's New Drugs and Clinical Trials Rules, 2019, which has substantially streamlined the governance of clinical trials. The article also discusses how AI is increasingly gaining importance in oncology trials. AI helps to design trials, improves the accuracy of clinical trial data, and promotes adaptive trial designs. AI aids recruitment by using electronic health records and genomic data to address major recruitment challenges. Besides allowing for concurrent real-time monitoring of safety and efficacy, such technologies can enhance speed in detecting adverse events and treatment outcomes. Through comparison of the regulatory practices and recognition of technological advances, this publication assists sponsors, CROs, and researchers to conduct oncology clinical trials that are more efficient, ethical, and globally harmonized.
Globally, cancer is the leading cause of morbidity and mortality. The incidence of cancer varies by area in India, making prevention and management extremely difficult. India's cancer burden was estimated to be 26.7 million DALYsAMI in 2021 and is predicted to rise to 29.8 million in 2025. The country's north and northeast had the highest burdens, with 2408 and 2177 DALYsAMI per 100,000, respectively, and among males. The seven most common cancer sites are lung (10.6%), breast (10.5%), esophagus (5.8%), mouth (5.7%), stomach (5.2%), liver (4.6%), and cervix uteri (4.3%) which accounts for over 40% of the overall cancer burden.[1]
Similarly, Cancer is also the second leading cause of death in the United States overall and the leading cause among people younger than 85 years. In 2025, 2,041,910 new cancer cases and 618,120 cancer deaths are projected to occur in the United States. Native American and Black people have two to three times the cancer death rate as White people for numerous cancers, many of which are largely preventable.[2]
The regulatory landscape for clinical trials is crucial for the development and approval of oncology products. Given the high prevalence of cancer and the continuous need for innovative treatments, understanding the regulatory requirements in major markets like India and the USA is essential for pharmaceutical companies and researchers. The regulatory frameworks in both India and the USA are complex and differ considerably. Understanding these variations is crucial for designing and executing successful clinical trials. Furthermore, regulatory approval in both markets is key to ensuring that new oncology treatments are accessible to a broader patient population. Oncology products represent a significant portion of the pharmaceutical market, contributing around 20% of global pharmaceutical sales. In India, oncology drugs make up roughly 12% of the pharmaceutical market, while in the USA, this figure rises to 25%.
India has increasingly become the preferred destination for clinical trials due to its diverse population and cost-effective trial environment. At the same time, the USA has a more stringent regulatory framework governed by USFDA, still each country ensures patient safety, data integrity, and scientific validity. While several publications discuss general clinical trial processes, this review analyzes and contrasts the regulatory authorities, safety reporting system, ethical oversight mechanisms, approval timelines involved in oncology trial in both countries. The objective is to understand the clinical trial requirements for oncological products in India, to identify the ethical guidelines for conducting cancer clinical trials, to create framework for the clinical trial regulatory approval processes for oncology trials in India and USA, to assess the differences in timelines and documentation requirements for clinical trials in India and USA, this will help researchers, sponsors and regulators in aligning global practices and enhancing the efficiency and ethical conduct of cancer clinical trial .
Drug development for oncology is a difficult, time-consuming, and risky process. Clinical trials frequently encounter difficulties with patient recruitment, trial design, data management, and outcome prediction. Artificial intelligence has become a game-changing tool in the biomedical industry in recent years, providing various ways to optimize different aspects of clinical trials. AI has the ability to greatly increase the development process of cancer drugs, from improving trial monitoring and adaptive designs to improving patient selection through predictive modeling. The use of AI in oncology clinical trials is examined in this review, with an emphasis on how it can boost productivity, cut expenses, and ultimately improve patient outcomes.
DISCUSSION:
Before a drug is offered for sale, it has to be approved, and a clinical trial has to be performed to ensure its safety and effectiveness. For the protection of the safety of the patients and ethical practice of clinical trial, the countries have provided clinical trial regulations, which are to be followed by the concerned organizations.
In terms of Clinical Research, particularly oncology trials, it is essential to understand the key stakeholders and concepts involved.
CLINICAL RESEARCH
“Clinical research” refers to studies, or trials, which are done in people. Clinical research is the key to the discovery of latest diagnostic methods and to develop modern drugs for treatment of diseases. Researchers design clinical trials to answer specific research questions related to a medical product. Before a clinical trial begins, researchers review prior information about the drug to develop research questions and objectives.
Clinical trials follow a typical series from early, small-scale, Phase 1 studies to late-stage, large scale, Phase 4 studies as follows,

Figure 1:Phases of Clinical trial
Phase 1 is termed the non-therapeutic phase,
Phase 2 is called exploratory trial phases,
Phase 3 is known as the therapeutic confirmatory phase, and
Phase 4 is called the post-approval or the post-marketing surveillance phase.[3]
Phase 0, also called the micro-dosing phase is specially perform for oncology study which was previously done in animals but now it is carried out in human volunteers to understand the dose tolerability (pharmacokinetics) before being administered as a part of the phase 1 trial among healthy individuals.[4]
There are three types of Phases 0 clinical trials:
The microdose Clinical trials is used to determine drug pharmacokinetics, to establish pharmacologically relevant doses and to study the mechanisms of action. Micro-dose trials are designed to evaluate pharmacokinetics, bioavailability, metabolism and/or distribution of a drug or its metabolite. This helps to determine the properties of two or more structurally similar analogues aimed at the same molecular target.
The second type of Phase 0 trials determine a dose regimen for molecular targeted compounds or bio-modulators meant for use along with other drugs like conventional chemotherapeutic agents. They do not determine the maximum tolerated dose, but they can be designed to determine a sequence of administration and a dose-range for subsequent combination trials.
The third type of Phase 0 trials, relate the mechanisms of action of drugs to drug efficacy; the pharmacodynamic effect of a drug can thereby be evaluated. It especially holds significance in the field of oncology for the evaluation of molecular targeted chemotherapeutic drugs.[5]
CLINICAL TRIAL DESIGNS:
“Clinical trial design refers to the framework or methodology used to conduct clinical research in a structured and scientifically valid way”. It defines how a clinical study is planned, including the selection of participants, the intervention or treatment being tested, the controls used for comparison, and the way data is collected and analysed. The primary goal of trial design is to ensure that the study provides reliable results while safeguarding patient safety and minimizing bias.
Clinical research design are of two major types that include non-interventional/observational and interventional/experimental studies. The non-interventional studies may have a comparator group (analytical studies like case-control and cohort studies), or without it (descriptive study). The experimental studies may be either randomized or non-randomized. Clinical trial designs are of several types that include parallel design, crossover design, factorial design, randomized withdrawal approach, adaptive design, superiority design, and non-inferiority design as follows:
TYPES OF RANDOMIZATIONS IN CLINICAL TRIAL:
In clinical trial Randomization is used to avoid bias, the types of randomizations are as follows:
CLINICAL TRIAL METHODS: -
The clinical trial methods are as follows:
DIFFERENCE BETWEEN ONCOLOGY CLINICAL TRIAL AND OTHER CLINICAL TRIAL: -
Oncology clinical Trial differs from other non-oncology trials in many ways. Most notably, cancer is understood as many diseases, not just one. For instance, types of cancer can include lung cancer, breast cancer, kidney cancer and many more. This makes oncology clinical trials particularly complicated in comparison to other therapeutic areas. The difference is as follows:
The endpoints, for oncology clinical trial and other clinical trial differ greatly, for example, rather than running a clinical trial to test the safety and efficacy of an antibiotic against an infection, an oncology trial is trying to extend and improve a subject’s quality of life. The endpoints in oncology clinical trial are overall survival which is considered as the “Gold standard” primary clinical endpoint, Progression-free survival , Time to progression , Event-Free Survival , Disease-free survival , Time to Treatment Failure , Time to Next Treatment , Duration of Clinical Benefit , Duration of Response, Objective Response Rate , Complete Response, Pathological Complete Response , Disease Control Rate , Clinical Benefit Rate , Health-Related Quality of Life , Milestone survival .[7]
In the typical clinical trial process, a placebo is used in place of the treatment to measure the effectiveness of new development therapies in comparison, whereas in case of oncology clinical trial Placebos are never used in place of treatment when an existing standard therapy exists. If a patient is given a placebo in an oncology trial, it is always in conjunction with other approved treatments.
In non-oncology trials, adverse events are measured as mild, moderate or severe. However, in oncology clinical trials, adverse effects are measured using a unique grading system, which rates events from 1 to 5 depending on severity.
These guidelines are administered by the National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE). These oncology-specific guidelines classify adverse events as follows:
The full criteria may not apply to all adverse events. For example, iron overloads are classified as level 2 adverse events at a minimum, whereas events including tinnitus and vertigo are only classified as high as level 3.
Patient recruitment is often more of a challenge with oncology trials. Often more sites are needed to meet population requirements which increases costs to the sponsor incrementally. According to Applied Clinical Trials, Lack of participation can cause an oncology trial to recruit slowly, often lengthening the trial's timeline by months or even years.[8]
ETHICS COMMITTEE:
The Role of ethics committee is to monitor all the protocols of the research study, the informed consent form, and other necessary documents that are related to the research proposal. It is the responsibility of the ethics committee to give approval of the study after reviewing all documents to monitor the study on a daily basis and also maintain the confidentiality of the records. It is the duty of ethics committed to protect the rights and safety of patients involved in study. All records must be maintained for 5 years after completion of the study. This also examines Severe adverse events and report it to DCGI.[9]
SPONSORS:
“The sponsor has the responsibility for starting, managing and giving financial support to a clinical trial”. The investigator can also play the role of Sponsor. The sponsor can transfer the trial related activity to other CRO or scientific body, but it should be documented in written. The sponsor must have knowledge for conducting trial, it must evaluate the investigator before starting clinical trial. The investigator Brochure must be provided to investigator, and all details of investigator must be verified. It is the responsibility of the Sponsor to obtain approval from DCGI and ethics committee. The sponsor has to submit the Protocol Report, Case Report form and ICF.[9]
INVESTIGATOR:
The role of investigator is to Conduct and supervise the whole study. The investigator must have the required qualification, experience and training as per the protocol. Before starting the trial, the investigator and sponsor must have agreement on paper signed, monitoring responsibilities etc. The work of investigator is to obtain Informed consent from patients, approval from ethics committee, apart from that any adverse event reported must be reported within the provided time. Investigator cannot take more than three trials at the time.[9]
CLINICAL TRIALS REGISTRY-INDIA: -
This is free, searchable online site where clinical trial registration can be done which are performed in India. All types of clinical trial can be registered here e.g. Observational data, interventional trial, Phase IV study, etc. These are easy to search and handle by common people from its official page. The World Health Organization reorganize the CTRI as the primary registry in the year 2008, from this year every month data gets transferred from CTRI to the international clinical trials registry site. In year 2009, CDSCO made it compulsory for every clinical trial to be register in CTRI.[9]
PROTOCOL: -
“Protocol is a document which provide the background, objectives, rationale, design, methodology (including the methods for dealing with adverse events, withdrawals etc.) of the study”. It also provides the criteria in which the study will be performed. The protocol must provide detailed processes for carrying out the trial, to ensure that the personnel coming in contact with the clinical trial collect data as per the laws. If any amendments are to be made in protocol, then it should be notified and approved by CLA, along with approval from Ethics Committee.[10]
INVESTIGATOR BROCHURE: -
“Investigator Brochure is a collection of data which includes justification for the proposed study for the Investigator consisting of all the clinical as well as non-clinical information available on the Investigational Product known prior to the onset of the trial”.
The IB should include Sponsor’s name, the reference number allocated to the study, the identity of each investigational product (i.e. research number, chemical or approved generic name, and trade name where legally permissible and desired by the sponsor).
The IB should bear an edition number and date. wherever applicable it also bears a reference to the number and date of supersedes version.[11]
INFORMED CONSENT: -
Informed Consent is an important part of any ethical research work. In all study research written informed consent is to be obtained from each study subject. The subject must be informed verbally about the type of study, risks, benefits etc. in easy language that is understandable to participants. It should be approved by the ethics committee and furnished by the licensing authority. If a person is not able to give informed consent, then a legally authorized representative should provide consent on its behalf.[12]
APPROVAL PROCESS FOR CONDUCTING CLINICAL TRIAL IN INDIA:
Step 1: Approval from the Drugs Controller General, India.
Step 2: Permission from particular Ethics Committee where the clinical study is designed.
Step 3: Compulsory registration on the ICMR website www.ctri.in. [13]
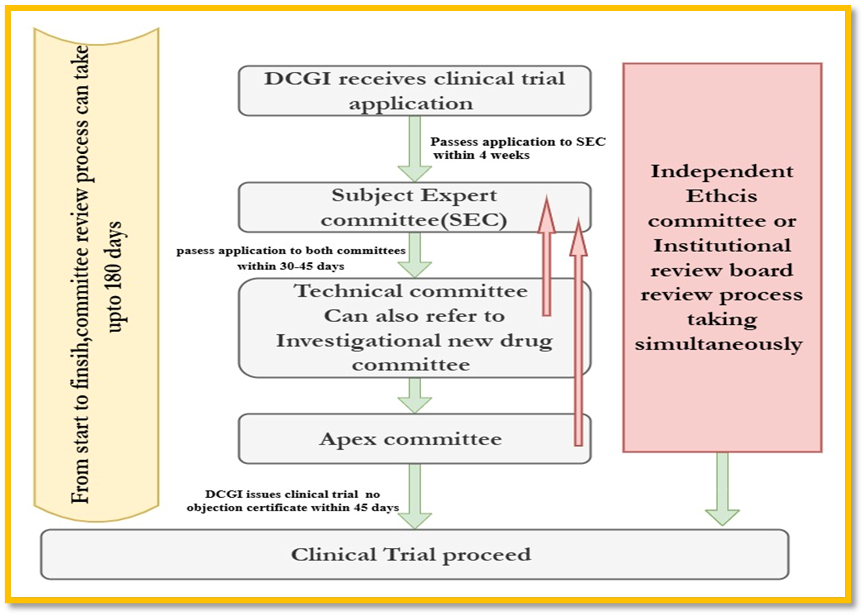
Figure 2:Clinical Trial Approval process in India
APPROVAL PROCESS FOR CONDUCTING CLINICAL TRIAL IN USA:
If the drug from the Preclinical trial is found safe, then Clinical Trial in Humans need to be performed before getting approval for marketing drugs. The Investigational New Drug Application is an application filed to FDA to start clinical trials in the USA. It is the Responsibility of the Sponsor to file Investigational New Drug application. The Food & Drug Administration does not charge any fee to review investigational new drug submissions.[14]
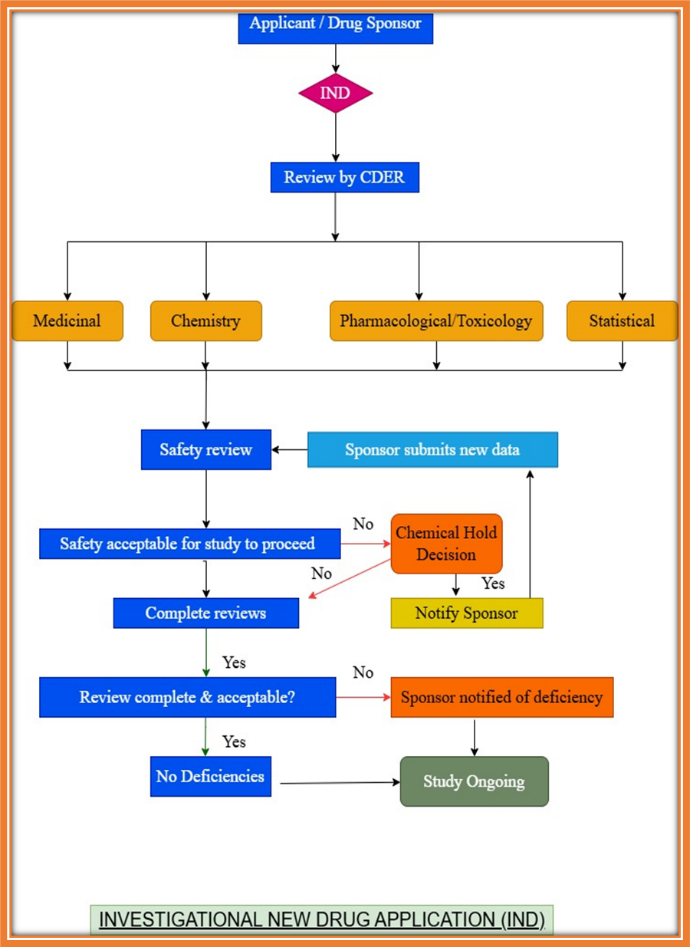
Figure 3:Clinical trial approval process in USA
COMPARISON CLINICAL TRIAL APPROVAL PROCESS REQUIREMENTS IN INDIA AND USA [15]
Table 1:Comparison of Clinical Trial process in INDIA and USA
|
SR. NO. |
PARAMETERS |
INDIA |
USA |
|
1 |
Regulatory Agency/Regulatory Authority Body |
DCGI, CDSCO |
USFDA |
|
2 |
Clinical Trial Application |
Application in CT-04, and Approval in CT-06 |
Investigational New Drug application |
|
3 |
Mandatory Documents Submission |
Dossiers, ICF, IB, CRF, CTA, diagrammatic flow chart for protocol. |
Dossiers, ICF, IB, CRF, CTA, diagrammatic flow chart for protocol. |
|
4 |
Clinical Trial registration Authority |
Clinical Trial Registry-India |
U.S national library of medicine under national institute of health |
|
5 |
Website |
http://CTRI.nic.in |
ClinicalTrials.gov, |
|
6 |
Registration method |
One-time registration |
One-time registration |
|
7 |
Payment For registration |
Not Required |
Not Required |
|
8 |
Document submission format |
Paper |
e-CTD, Paper |
|
9 |
Regulatory Authority Fees according to clinical trial Phase |
||
|
Phase I |
3,00,000 |
If relevant |
|
|
Phase II |
2,00,000 |
If relevant |
|
|
Phase III |
2,00,000 |
If relevant |
|
|
Phase IV |
2,00,000 |
70,480 |
|
|
10 |
Timelines for Protocol Review by Regulatory |
45 days |
Within 30 calendar days of receipt of the original IND |
|
11 |
Timelines for Protocol Review by IEC/IRB |
30-60 days |
30-45 days |
|
12 |
Fees required by ethics committee |
Required |
Required |
|
13 |
Regulatory guidelines |
New drugs and clinical trial rules, ICMR guidelines, Indian GCP. |
US-ICH-GCPs, FD & C act rules, Office of Good clinical practice |
|
14 |
SAE reporting Timeline |
24 hours |
As soon as possible no later than 7 calendar days |
|
15 |
Number of Protocol to be submitted |
4 copies like 2 hard copies 2 soft copies |
2 copies both e-CTD and PAPER |
|
16 |
Informed consent |
Must be in regional language, audio-visual recording mandatory for vulnerable populations |
Must be in plain English; FDA requires concise key information summary |
COMPARISON BETWEEN INSTITUTIONAL ETHICS COMMITTEE AND INSTITUTIONAL REVIEW BOARD:[15]
Table 2:Comparison between IEC and IRB
|
Sr. No. |
Features |
Institutional Ethics Committee |
Institutional Review Board |
|
1 |
Governing Guidelines |
ICMR, New Drugs and clinical trial rules under CDSCO |
Regulated by U.S. FDA (21 CFR Parts 50 & 56) and US department of health and human service (45 CFR 46) |
|
2 |
Jurisdiction |
India |
USA and internationally |
|
3 |
Focus |
Ethics in Indian clinical trials |
Ethics in global medical research |
|
4 |
Members |
Minimum 7 members: medical scientist, clinician, legal expert, social worker, lay person, etc. |
Minimum 5 members including: scientist, non-scientist, and someone unaffiliated with the institution |
|
5 |
Compensation for Trial-Related Injury |
Mandatory compensation as per NDCT Rules |
Compensation policies vary by sponsor/IRB; not mandated by law unless negligence is involved |
|
6 |
Registration
|
Mandatory registration with CDSCO (renewable every 5 years) |
Must register with Office for Human Research Protections and FDA, where applicable |
ARTIFICIAL INTELLIGENCE IN ONCOLOGY TRIALS:
Traditional oncology trials often face problems like strict protocols, slow patient recruitment, and diverse populations. These issues make it hard to evaluate targeted therapies. AI offers a chance to solve these challenges. It allows for data-driven, flexible, and personalized trial methods. In recent years, Artificial Intelligence has become a valuable tool in oncology clinical trials. It provides new ways to improve trial design, patient selection, data analysis, and execution.
AI systems, especially those which uses machine learning and deep learning algorithms, can analyze large and complex datasets, such as genomic profiles, histopathological images, electronic health records, and real-time patient monitoring data. These abilities enable AI to study detailed patterns and predict treatment responses in ways that weren't possible before. By grouping patients more effectively and connecting them to the best interventions, AI improves trial design, speeds up patient recruitment, and fine-tunes outcome assessment.
Furthermore, AI can significantly improve adaptive trial designs by allowing real-time changes based on collected data. For example, AI can adjust inclusion criteria, cohort sizes, or even intervention arms to reflect new efficacy signals. This not only will decrease the duration and cost of trials, but it will also make them more ethical by minimizing exposure to the treatments which are not effective. Also, AI helps to identify biomarkers and surrogate endpoints that can signal early signs of treatment success, that provide regulators and sponsors with faster paths to approval.
Researchers and medical professionals can make sure that the appropriate patients receive the right treatments at the right time by using AI to plan, carry out, and evaluate clinical trials.[16]
FUTURE OF AI IN ONCOLOGY TRIALS:
With continuous development of AI technologies and increasing accessibility to multi-modal cancer data, it helps in implementing clinical trials that are more efficient, inclusive, and effective. For example, the advancement of natural language processing allows trial datasets to integrate unstructured clinical notes, pathology reports, and literature, thereby vigorously expanding the scope of data-driven insights. On the side of multi-omics integration, giving insights from genomics, proteomics, and metabolomics such evidence may help identify new and previously inaccessible biomarkers and therapeutic targets.
Apart from this, the increased adoption of decentralized and hybrid trial models supported by AI-based telemedicine and remote monitoring solutions shall widely render clinical research acceptable by the patients. Such approaches lower the barriers to entry while enhancing trial diversity, thus yielding broadly applicable results. Machine-learning models, trained on data from these decentralized trials, can also better represent real-world treatment patterns and outcomes, thereby further bridging the gap between clinical research and day-to-day practice.
The fast-changing developments in technology will possibly be integrated into oncology trials with an increasing number of advanced techniques such as reinforcement learning strategies, augmented-reality tools for clinician support, and even AI-driven autonomous trial protocols that modify themselves instantaneously based on the outcomes of the procedure. The ultimate ambition is to set an evolving cycle whereby patient interactions will incrementally inform and teach us more about cancer biology and better therapies.[16]
ROLE OF AI IN TRIAL DESIGN, OPERATIONS, AND DATA MANAGEMENT:
AI can be used to change both interventional and non-interventional clinical trial designs. AI could indeed help in patient selection in clinical trial designs to define best target populations. The early detection could provide endpoints of studies or safety signals that allow adaptive trial design, as the trial would now be modified in support of the patient safety and outcome targets. Further, it would enhance the supposed application of machine learning-powered predictive models for deeper understanding of the intended-use population for patient stratification before any randomized assignment. Technologies are currently available to estimate how an inclusion criterion might modify trial outcomes using aggregated published trial results and to enter eligibility criteria to produce an appropriate patient database through web-based interfaces.
AI can enhance the representativeness of clinical trials by balancing cohorts with specific patient characteristics relevant to disease epidemiology. AI models are currently researching virtual patient simulations, or digital twins, of enrolled patients that may offer modeling approaches to increase statistical power and patient representation; using these specific approaches in the process of developing new drugs is still in the early stages. Besides patient selection, Machine learning based models can also gather and integrate large data sets generated by digital health technologies, like wearable devices can capture data on vital signs, physical states, pharmaceutical adherence, and other behavioral parameters that may inform patient outcomes. Such technologies and other decentralized components of oncology trials face considerable barriers, including enormous logistical challenges involved in shipping and administering trial medications, as well as coordinating clinical services across multiple geographic locations.
Artificial Intelligence and Machine Learning approaches may be applied in clinical trial data management and analysis for functions such as standardization of unstructured clinical data; harmonization of multiple dependent variables, trial sites, or institutions; and the de-identification of personal health information for enhancing privacy protection. AI can also enable pharmacometrics analysis on big data sets, for instance, identifying the optimum drug dose, drug or drug-food interactions, and pharmacokinetic variability across subgroups. To ensure the quality of collected data, machine learning techniques have been proposed to help in the identification and removal of duplicate participants, imputation of missing data points, and validation of novel data sets against existing reference databases. These applications are able to handle voluminous multidimensional internal missingness records, which is a common limitation in real-world data, but specific expertise in epidemiology, biostatistics, and data science is required to ensure its correct application.
Effective automation of some trial components will increase efficiency and minimize the burden on investigators. Ambient AI scribes demonstrate promise for outpatient settings by decreasing clinical documentation time and enhancing physician engagement. AI scribes may summarize subjective reports, pend orders, and generate treatment plans within the protocol for clinical trials. LLMs can produce personalized patient education materials that may be written in the patient's native language. LLMs that are commercially available for most doctors offer compliant document generation regarding HIPAA. AI technologies will be integrated into many current clinician workflows with clinician supervision, from cohort definition to the clinic visit.[17]
CONCLUSION
This review provides the critical differences and emerging alignments in the clinical research requirements for oncology products in India and the USA. While the FDA provides a mature and comprehensive regulatory framework in the U.S., India’s CDSCO is progressively evolving its processes to meet global standards. Despite ongoing improvements, India faces unique challenges such as limited infrastructure, regulatory delays, and patient recruitment barriers that can impact oncology drug development timelines.
With opportunities for transformation across two regulatory landscapes, artificial intelligence in oncology clinical trials is an entirely new opportunity set. From optimizing trial design through patient stratification and real-time data analysis, AI could increase trial efficiency and probability of success significantly. Implementation, however, is still in infancy issues regarding data privacy, validation of algorithms, and regulatory clarity remain unresolved. It is imperative that regulators work together with researchers and technology developers to ensure that AI can be best utilized in an ethically and clinically relevant manner.
ACKNOWLEDGMENT
I would like to express my sincere gratitude to Ms. Grishma Desai, for her valuable guidance, encouragement, and insightful feedback throughout the preparation of this review article. Her support has been instrumental in shaping my understanding of the subject and in refining the quality of this work. I am deeply thankful for the time and effort she gave in mentoring me.
CONFLICTS OF INTEREST STATEMENT:
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
REFERENCES


Sonal Singh, Grishma Desai, Vineet Jain, Comparative Analysis of Clinical Research Requirements for Oncological Products in India and USA with Emerging AI Applications, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 12, 2152-2165. https://doi.org/10.5281/zenodo.17912055
 10.5281/zenodo.17912055
10.5281/zenodo.17912055