
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1Dean, Faculty of Medical Science and Research, Sai Nath University, Ranchi, Jharkhand 835219, India.
2Assistant Professor, Department of Pharmacy, Sai Nath University, Ranchi, Jharkhand 835219, India.
3Student, B. Pharm, Department of Pharmacy, Sai Nath University, Ranchi, Jharkhand 835219, India.
4Assistant Professor of Pharmacology, Department of Pharmacy, Faculty of Medical Science and Research, Sai Nath University, Ranchi, Jharkhand 835219, India
This comprehensive review explores the complex neurobiological underpinnings of anxiety disorders and the challenges associated with developing effective pharmacological treatments. The article examines the roles of key neurotransmitter systems, including serotonergic and GABAergic pathways, in anxiety regulation. It discusses the involvement of the hypothalamic-pituitary-adrenal (HPA) axis and its dysregulation in anxiety disorders. The review also addresses the molecular mechanisms underlying the therapeutic effects of antidepressants and anxiolytics, with a focus on the adenylate cyclase-cAMP-protein kinase A signaling cascade and its impact on gene transcription. Additionally, it explores the potential of novel targets, such as neuropeptide systems and glutamatergic receptors, in anxiety treatment. The article highlights the limitations of current preclinical models and the difficulties in translating promising compounds into clinically effective therapies. It concludes by emphasizing the need for a multidisciplinary approach to overcome the challenges in anxiety drug development and to address unmet medical needs in this field.
Anxiety disorders encompass a diverse group of psychiatric conditions marked by excessive fear or worry, contributing significantly to public health concerns due to their high prevalence and associated comorbidities. Despite substantial research efforts, the precise neurobiological mechanisms underlying these disorders remain poorly understood, which impedes the development of targeted pharmacological treatments.[1]
The DSM-5 (Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition) classifies anxiety disorders into various subtypes, each presenting distinct clinical features. However, the absence of specific biomarkers complicates the accurate diagnosis and hampers the advancement of tailored therapies.[2] Although the serotonergic and GABAergic neurotransmitter systems have been implicated in the pathophysiology of anxiety, the exact mechanisms by which these systems influence the development and progression of these disorders are still not fully clarified.[3]
Chronic stress plays a pivotal role in the onset and persistence of anxiety disorders, primarily through the dysregulation of the hypothalamic-pituitary-adrenal (HPA) axis—a critical component of the body's stress response system.[4] This dysregulation, often characterized by hyperactivity, leads to elevated cortisol levels and disrupts the function of brain regions involved in emotional processing, such as the amygdala, hippocampus, and prefrontal cortex.[5,6]
The amygdala, a central structure in the brain's fear circuitry, is crucial for the acquisition and expression of fear and anxiety. Individuals with anxiety disorders frequently exhibit hyperresponsivity in the amygdala in response to threatening stimuli. [7,8] In contrast, the hippocampus, which plays a vital role in memory formation and learning, often shows reduced volume and functional impairments in those with anxiety disorders.[9] The prefrontal cortex, responsible for executive functions and regulating emotions, also experiences structural and functional changes that contribute to anxiety symptoms.[10]
The complex interactions among these brain regions, coupled with HPA axis dysregulation, are integral to the development and persistence of anxiety disorders. [11] While current treatments, such as selective serotonin reuptake inhibitors (SSRIs) and benzodiazepines, can alleviate symptoms for many patients, there is a pressing need for new therapeutic strategies that address the underlying biological mechanisms.[12]
To enhance our understanding of anxiety disorders and improve treatment outcomes, future research must focus on unraveling the intricate relationships between genetic predispositions, environmental stressors, and neurobiological processes.[13] Additionally, the identification of specific biomarkers for different anxiety subtypes will be essential for advancing personalized medicine. By overcoming these challenges, the development of targeted interventions may lead to significant advancements in the management of anxiety disorders, ultimately improving patient care and quality of life. [14]
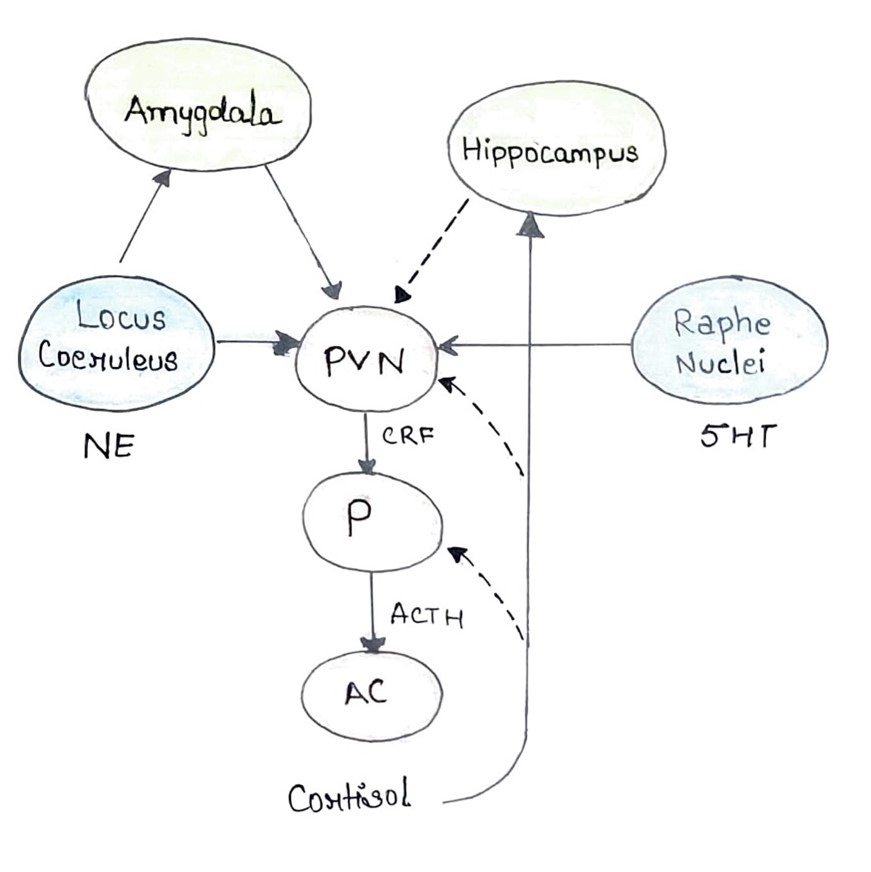
Fig. 1. The hypothalamic-pituitary-adrenal (HPA) axis governs the body's response to stress through a series of interconnected processes. Initiated in the hypothalamic paraventricular nucleus (PVN), corticotropin releasing hormone (CRH) is released into the hypophyseal portal blood, traveling to the pituitary gland (P). Here, adrenocorticotropic hormone (ACTH) is synthesized and released into systemic circulation, ultimately reaching the adrenal cortex (AC). Cortisol, the end product, is then synthesized and released into the bloodstream. Regulation of the HPA axis involves complex neural interactions. The amygdala provides stimulatory projections, while the hippocampus offers inhibitory influences. Additionally, the PVN receives noradrenergic projections from the locus coeruleus (LC) and serotonergic projections from the raphe nuclei (RN). Feedback loops play a crucial role in modulating this system. Cortisol exerts negative feedback by inhibiting ACTH synthesis in the pituitary and CRH synthesis in the PVN. It also stimulates the hippocampus, which further inhibits the PVN. Under conditions of chronic stress, these feedback mechanisms can become dysregulated, leading to hypercortisolism. This state is accompanied by heightened amygdala reactivity and reduced hippocampal function.Effective treatment aims to restore these feedback mechanisms, resulting in normalized cortisol levels, reduced amygdala reactivity, enhanced hippocampal function, and balanced noradrenergic and serotonergic activity.
Role of Serotonin Transporter in Anxiety
There are at least 14 distinct serotonin receptor subtypes, including 5-HT1 (A, B, D, E, F), 5-HT2 (A, B, C), 5-HT3, 5-HT4, 5-HT5 (A, B), 5-HT6, and 5-HT7. All of these receptors, except 5-HT3, are G-protein-coupled receptors.[15] The signaling pathways mediated by these G-protein-coupled serotonin receptors are highly diverse, and the specific functions and interactions of these various receptors within the central nervous system (CNS) remain largely unknown.[16,17] This complexity, coupled with the widespread distribution of serotonin receptors throughout the brain, positions the serotonergic system as a critical modulator of various brain functions. Disruptions in serotonergic neurotransmission have been linked to altered vulnerability and the development of psychiatric conditions, including depression, anxiety disorders, and schizophrenia.[18]
All serotonin receptors are located postsynaptically on non-serotonergic neurons, referred to as heteroreceptors. However, 5-HT1A and 5-HT1B/1D receptors also function as autoreceptors on serotonergic neurons,[19] with 5-HT1A found on the soma and 5-HT1B/1D located in the synaptic area. The serotonin transporter (5-HTT) is found in both the synaptic region and the cell bodies of serotonergic neurons.[20] Upon neuronal firing, serotonin is released into the synaptic cleft, where it acts on nearby pre- and postsynaptic serotonergic receptors. The 5-HTT plays a key role in regulating serotonin signaling by reuptaking serotonin from the extracellular space back into the neuron, which terminates its signaling and allows serotonin to be recycled for future release through uptake by the vesicular monoamine transporter (VMAT2).[21,22] Another mechanism for ending serotonergic activity involves the uptake of synaptic serotonin by surrounding glial cells, where it is degraded into its main metabolite, 5-hydroxyindole acetic acid (5-HIAA) by monoamine oxidase-A (MAO-A).[23] Selective serotonin reuptake inhibitors (SSRIs) are currently the first-line pharmaceutical treatment for anxiety disorders, while serotonin-norepinephrine reuptake inhibitors (SNRIs) are considered second- or third-line options.[24] A meta-analysis comparing the efficacy of SSRIs, SNRIs, and placebos in treating anxiety disorders, depression, obsessive-compulsive disorder (OCD), and post-traumatic stress disorder (PTSD) in children and adolescents found that SSRIs and SNRIs were similarly effective and superior to placebo.[25;26] Despite the notable placebo effect commonly observed in psychiatric treatments, SSRIs' inhibition of 5-HTT in the CNS is widely recognized to have anxiolytic effects across diverse patient populations with anxiety and fear-related disturbances.[27] The involvement of the 5-HTT in anxiety mechanisms has been extensively studied. Inhibiting 5-HTT results in an immediate increase in synaptic serotonin concentration; however, the anxiolytic effects typically emerge only after prolonged treatment, suggesting that long-term plasticity changes in anxiety mechanisms are necessary.[28,29] The specific mechanisms underlying these effects remain largely unknown, but it is evident that the pathogenesis of anxiety disorders, particularly generalized anxiety disorder (GAD), is influenced by numerous factors, including complex interactions between biological, environmental, and psychological elements.[30,31] Genome-wide association studies (GWAS) and linkage studies in large populations with anxiety disorders have not consistently identified associations with the 5-HTT. However, candidate gene studies have provided substantial evidence for the serotonergic system's role in anxiety-related endophenotypes. [32] Polymorphisms in the 5-HTT gene, including its associated transcriptional regulatory regions, have been shown to influence serotonergic activity. The 5-HTT gene, located on chromosome 17 (17q11.2), spans approximately 40 kb and comprises 14 exons, encoding a protein with 12 transmembrane domains.[33] Variants in the 5-HTTLPR promoter region, such as the S- and L-alleles, and other polymorphisms, influence 5-HTT expression and function. Individuals with genotypes associated with lower 5-HTT expression may experience greater 5-HTT occupancy and saturation when treated with SSRIs, potentially leading to increased central and peripheral serotonin levels and associated side effects.[34] The 5-HTT is a crucial modulator of the serotonergic system, with its activity significantly influencing the duration and magnitude of serotonergic neurotransmission. Dysregulation of 5-HTT signaling pathways has been linked to various psychiatric disorders, including anxiety. [35]Initial research suggested that the S- and L-variants of the 5-HTTLPR might differentially influence vulnerability to neuropsychiatric disorders or predict responses to SSRIs. However, recent findings suggest a more complex pattern, with multiple polymorphisms within the promoter region affecting 5-HT reuptake properties.[36] The modulation of serotonergic transmission via 5-HT mechanisms highlights the complexity of genetic and functional interactions involved in psychiatric disorders. The identification of specific gene variants associated with anxiety has proven challenging, as these variations likely interact with other genetic, epigenetic, environmental, and developmental factors to influence the risk or resilience to psychiatric conditions.[37] Using intermediate phenotypes may facilitate the identification of associations between specific candidate genes, such as 5-HTT, and complex psychiatric phenotypes. Various studies are exploring gene-environment and gene-gene interactions in the 5-HTTLPR promoter region, with findings suggesting that the S-allele is associated with increased risk for psychiatric disorders, including anxiety.[38] The role of the L-allele is less clear, although it may also contribute to the development of psychopathological traits. Given the ubiquity of these alleles and the fact that not everyone develops psychiatric disorders, it is likely that protective alleles, such as certain variants of the CRF1 receptor, contribute to individual resilience.[39] Overall, while genes certainly play a role in the development of anxiety disorders and related traits, no single gene has been identified as the primary contributor to vulnerability. Meta-analyses of genotypes linked to mental disorders with varying degrees of heritability suggest that only a few genes are associated with anxious phenotypes, in contrast to the numerous genes implicated in disorders like schizophrenia.[40] The complexity of these findings underscores the challenges in identifying new targets for anxiolytic drugs, as anxiety disorders likely result from disturbances in extensive neural networks and pathways influenced by multiple biological mechanisms.[41]
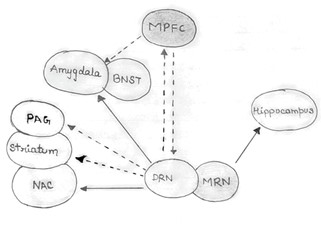
Fig. 2. The 5HT (serotonin) projections originating from the raphe nuclei (RN) have diverse effects on various brain regions, influencing behavior and emotional responses. The dorsal raphe nucleus (DRN) sends excitatory signals to the central nucleus of the amygdala (CeA) and the bed nucleus of the stria terminalis (BNST), both of which are involved in the regulation of fear and anxiety. It also sends inhibitory signals to the periaqueductal gray (PAG) and the striatum, which are associated with passive behaviors. Additionally, the DRN projects inhibitory signals to the medial prefrontal cortex (MPFC) and excitatory signals to the nucleus accumbens (NAc), influencing complex behaviors and emotional expression. In contrast, the MPFC sends inhibitory projections back to the DRN and the amygdala, potentially exerting an anti-anxiety effect. The medial raphe nucleus (MRN) projects excitatory signals to the hippocampus, which has been linked to increased resilience to stress and reduced anxiety levels. However, under conditions of chronic, uncontrollable stress, the DRN increasingly stimulates the amygdala, leading to heightened anxiety. Simultaneously, the DRN inhibits the PAG and striatum, promoting passivity, while serotonergic activity from the MRN to the hippocampus becomes impaired. This imbalance, characterized by increased amygdala reactivity and reduced hippocampal activity, can contribute to the overactivation of the hypothalamic-pituitary-adrenal (HPA) axis.
The Role of Animal Models in Understanding Anxiety and the Efficacy of SSRIs
Human linkage and association studies have yet to pinpoint genes that significantly influence the modulation of anxiety or anxiety disorders. Consequently, animal models are invaluable in exploring the genetic underpinnings of these conditions. Notably, anxiety-like behaviors observed in humans are also present in animals, providing a foundation for studying the neurobiological mechanisms involved.[42] Animal models have substantially advanced our understanding of the genetic and environmental factors contributing to anxiety. To further this research, a translational approach is essential, whereby findings from human studies inform animal research, and vice versa. Emerging technologies, such as cell-specific and inducible knockout models, optogenetics, and CRISPR/Cas9, enable precise manipulation of cellular mechanisms linked to gene function, and these techniques are increasingly being applied to animal models of fear and anxiety.[43] Over the years, numerous preclinical tests for anxiety have been developed, relying on face validity, predictive validity, and construct validity. However, no current models fully meet all these criteria. Many of these models, often using rodents like rats and mice, are based on natural behaviors and include tasks such as open field tests, elevated plus mazes, and light/dark box experiments.[44] These unconditioned and conditioned paradigms measure anxiety through avoidance behaviors and physiological responses. Research has also introduced models to investigate the role of the serotonin transporter (5-HTT) and adaptive serotonin (5-HT) signaling, particularly in the context of selective serotonin reuptake inhibitors (SSRIs). Findings from these studies have shown that 5-HTT overexpression or knockout in rodents can influence anxiety levels, shedding light on the complex interactions between 5-HTT gene variants and environmental stressors.[45] Despite the usefulness of these models, there remains variability in the efficacy of SSRIs across different tests and models, paralleling the inconsistencies observed in human clinical responses. While SSRIs are commonly used to treat anxiety disorders in humans, their effectiveness in animal models is inconsistent, particularly in acute studies.[46] This highlights the need for more chronic treatment models to better mimic the therapeutic effects observed in humans. Our own research aligns with this profile, showing that while SSRIs like fluvoxamine and clomipramine are effective in some behavioral paradigms, they are ineffective in others, underscoring the complexity of anxiety and its treatment.[47]
The Role of 5-HT1A Receptors in the Anxiolytic Activity of SSRIs and the Complexities of Anxiety Regulation
The anxiolytic effects of selective serotonin reuptake inhibitors (SSRIs) are partly attributed to the increased serotonin (5-HT) release resulting from the inhibition of the serotonin transporter (5-HTT). This enhanced 5-HT availability likely exerts its effects through interactions with various 5-HT receptors, with the 5-HT1A receptor (5-HT1AR) being a key candidate.[48] As early as 1979, it was discovered that buspirone, a partial 5-HT1AR agonist and dopamine D2 receptor antagonist, produced mild anxiolytic effects in patients with generalized anxiety disorder (GAD). These findings were later corroborated in rodent models of anxiety.[49] Although the clinical development of new 5-HT1AR agonists for anxiety disorders (e.g., ipsapirone, gepirone, tandospirone, flesinoxan) was ultimately unsuccessful, the 5-HT1AR remains a promising target in the treatment of anxiety and depression.[50] The 5-HT1ARs are inhibitory G-protein-coupled receptors that function as autoreceptors on serotonergic neurons and as heteroreceptors on non-serotonergic neurons, such as GABAergic neurons. The somatodendritic 5-HT1A autoreceptors play a crucial role in regulating serotonergic tone via feedback inhibition, working in concert with 5-HTT.[51] These receptors are predominantly located in the dorsal and median raphe nuclei, while postsynaptic 5-HT1ARs are highly expressed in specific brain regions, particularly in the limbic system, including the hippocampus and various areas of the frontal cortex.[52] Genetic and imaging studies have provided evidence that variations in 5-HT1AR density or regulation are associated with anxiety. For instance, a C(-1019)G polymorphism (rs6295) in the promoter region of the 5-HT1AR gene (Htr1a) has been linked to mood-related traits, such as amygdala reactivity. The G-allele is associated with increased 5-HT1A autoreceptor expression, while the C-allele is linked to enhanced postsynaptic expression. Variations in this polymorphism have also been found to influence defensive behavior, amygdala activity, and neural plasticity in patients with panic disorder and agoraphobia, suggesting a role for differential 5-HT1AR activity in anxiety and fear.[53] Griebel and Holmes summarized the effects of 5-HT1AR agonists in animal models of anxiety and fear, finding that approximately 65% of these agonists demonstrated anxiolytic activity, while 30% were inactive, and 5% were anxiogenic. Knockout studies of 5-HT1AR in different mouse strains consistently resulted in enhanced anxiety in standard anxiety tests, although the anxious phenotype was found to depend on the specific paradigm applied.[54] Notably, in one strain, but not others, 5-HT1AR knockout mice showed reduced sensitivity to the anxiolytic effects of diazepam, associated with changes in the GABAA-benzodiazepine receptor complex. However, this dysfunction of the GABAA-benzodiazepine complex is not required for the anxiogenic phenotype of 5-HT1AR knockout mice, and the increased anxiety in these mice could not be alleviated by SSRIs.[55] Conversely, overexpression of 5-HT1ARs reduced anxiety, providing strong support for the involvement of 5-HT1ARs in anxiety and fear processes. Further research has highlighted the differential roles of pre- and postsynaptic 5-HT1ARs in anxiety. For example, pharmacological blockade of 5-HT1ARs during early development, but not in adulthood, increased anxiety in normal mice. These findings emphasize the complex regulation of anxiety processes across different stages of development and adulthood, reflecting the intricate interplay between genetic factors, environmental influences, and neural circuitry.[56] While simple relationships between specific receptors, such as 5-HT1AR, and the regulation of anxiety and fear are unlikely, rodent models remain invaluable for disentangling the underlying mechanisms. In a preclinical research, both partial and full 5-HT1AR agonists exhibited anxiolytic activity. The azapirones (e.g., buspirone, ipsapirone, tandospirone, zalospirone) are partial 5-HT1AR agonists, while flesinoxan is a potent and selective full agonist.[57] However, 5-HT1AR agonists have shown limited anxiolytic effects in animal anxiety models based on the release of suppressed behavior. In contrast, these agonists are highly active in animal models of stress-evoked behaviors, such as fear-potentiated startle and stress-induced hyperthermia. [58]Although buspirone is marketed as an anxiolytic worldwide, and tandospirone is available in Japan and China, 5-HT1AR agonists have not emerged as a breakthrough in the treatment of anxiety disorders, failing to replace benzodiazepines as the standard treatment.[59]
Serotonin Receptor Modulation and Anxiety
The past three decades have witnessed a concerted effort to develop pharmacotherapeutic interventions for anxiety disorders by targeting the diverse serotonin (5-HT) receptor family. A plethora of agonists and antagonists selective for various 5-HT receptor subtypes have been synthesized and subjected to rigorous preclinical evaluation in anxiety-related behavioral paradigms.[60] Among the most extensively studied receptor targets are the 5-HT2A and 5-HT3 receptors. Meta-analyses of preclinical studies have consistently demonstrated anxiolytic-like effects for antagonists of both receptor subtypes. However, despite promising preclinical data, particularly for 5-HT3 receptor antagonists which were once heralded as potential breakthrough anxiolytics, clinical trials have failed to translate these findings into efficacious treatments for human anxiety disorders.[61] To date, the development of novel anxiolytic agents based on serotonin receptor modulation has yielded limited clinical success. This therapeutic gap underscores the complexity of anxiety disorders and the challenges associated with identifying and validating robust pharmacological targets within the serotonergic system. [62]
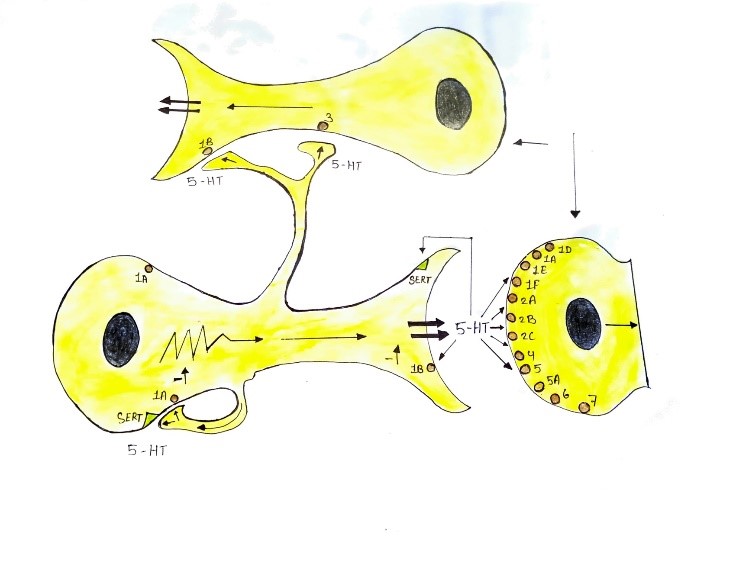
Fig. 3. This illustration depicts a serotonergic neuron interacting with two postsynaptic non-serotonergic neurons (one at the top and one on the right). The distribution of various 5-HT receptors and the serotonin transporter (5-HTT) on these neurons is outlined schematically. The 5-HT1A receptors are found both as somatodendritic autoreceptors on the serotonergic neuron and as postsynaptic heteroreceptors on the non-serotonergic neurons. The 5-HT1B/1D receptors are present as presynaptic receptors within the synaptic cleft and as postsynaptic heteroreceptors. The 5-HTT is located on the presynaptic region and soma of the serotonergic neuron. All other types of 5-HT receptors are situated postsynaptically on the non-serotonergic neurons.
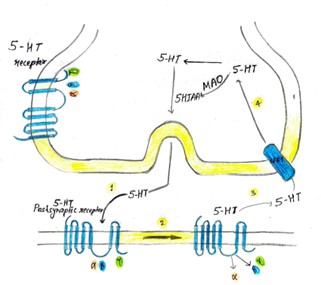
Fig. 4. This schematic illustrates the process of serotonin (5-HT) signaling at the synapse. G-protein-coupled receptors are found either presynaptically as 5-HT autoreceptors (5-HT1A/1B) or postsynaptically as various 5-HT receptors (types 1 through 7). The sequence of events is as follows: 1.Serotonin is released from the presynaptic neuron and binds to a postsynaptic receptor associated with a heterotrimeric G-protein. This G-protein complex consists of three subunits: ?, ?, and ?, with GDP bound to the ? subunit in its inactive state. 2. When serotonin binds to the postsynaptic receptor, it triggers a conformational change in the receptor. This change causes GDP to be replaced by GTP on the ? subunit, activating it, while the ? and ? subunits are released. 3. The extracellular serotonin is then reabsorbed into the presynaptic neuron via the serotonin transporter (5-HTT). 4. Once inside the presynaptic neuron, serotonin is either broken down by monoamine oxidase (MAO) into 5-hydroxyindoleacetic acid (5-HIAA), which can also occur extracellularly, or it is stored in vesicles for future release.
Key abbreviations include MAO (monoamine oxidase), 5-HIAA (5-hydroxyindoleacetic acid), and SERT (serotonin transporter).
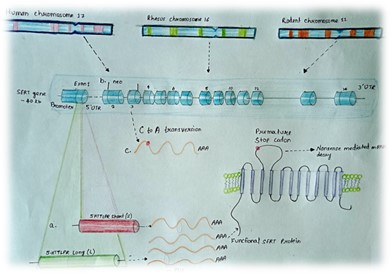
Fig. 5. This diagram illustrates various genetic alterations in the 5-HTT gene across humans, rhesus macaques, and rodents lead to differences in the transcription levels of the serotonin transporter (5-HTT). In humans and rhesus macaques, the presence of short or long alleles of the 5-HTTLPR polymorphism is associated with lower or higher transcription levels of 5-HTT, respectively. Rodents, however, lack an orthologous polymorphism. Instead, researchers can achieve a knockout of the 5-HTT gene in these animals through different methods:
(a) In mice, this is done by replacing exon 2 with a neo cassette, while (b) in rats, a premature stop codon is introduced in exon 3, both of which result in the loss of functional 5-HTT protein.
Role of GABAA Receptors in Anxiolytic Therapy and the Challenges of Benzodiazepine Use
GABAA (?-aminobutyric acid type A) receptors serve as the primary molecular targets for benzodiazepines (BDZ), a class of drugs discovered in the 1950s that became the cornerstone of anxiolytic treatment for decades. Beyond their anxiolytic effects, BDZs are also known for their sedative, anticonvulsant, muscle-relaxant, and hypnotic properties. However, it wasn't until over 20 years later that the molecular target of BDZs, the GABAA receptor (GABAA, R), was identified. These receptors are ionotropic inhibitory receptors and are integral to the pharmacological action of several clinically significant anxiolytics, including BDZs.[63] Despite their efficacy and safety in treating anxiety in both humans and animals, long-term BDZ use is associated with risks such as tolerance, physical and psychological dependence, and withdrawal symptoms upon discontinuation. Additionally, side effects like sedation and cognitive impairment limit their use. Consequently, selective serotonin reuptake inhibitors (SSRIs) and, to a lesser extent, serotonin-norepinephrine reuptake inhibitors (SNRIs) have become the first-line pharmacological treatments for many anxiety disorders.[64] BDZs are now typically reserved as second- or third-line options when antidepressants and cognitive-behavioral therapy prove ineffective. Nevertheless, BDZ prescriptions remain prevalent, with over 5% of adults in the United States being prescribed these drugs annually. GABAA receptors are widely distributed throughout the brain and are crucial for the precise timing of neuronal activity and the synchronization of neuronal networks.[65] The inhibitory neurotransmission mediated by these receptors is regulated by a diverse array of interneurons and the heterogeneous composition of GABA receptors themselves. GABAA receptors consist of five subunits arranged in a pentameric structure that surrounds a central pore. When GABA, the endogenous ligand, binds to the receptor, chloride ions flow into the neuron, leading to membrane hyperpolarization and inhibition of neuronal firing. GABAA receptors are localized both synaptically and extrasynaptically and exhibit significant molecular diversity due to the variable subunit composition.[66] The GABAA receptor subunits include ?1-6, ?1-3, ?1-3, ?, ?, ?, and ?1-3, with the majority of GABAA receptors being composed of two ?, two ?, and one ? subunit. The GABA binding site is formed by the ? and ? subunits, while the BDZ binding site is created by one of the ? subunits (?1, 2, 3, or 5) in combination with the ?2 subunit.[67] Approximately 60% of GABAA receptors are of the ?1?2?2 subtype, with other subtypes such as ?2?3?2 and ?3?n?2 making up smaller proportions. This subunit composition is not random and plays a crucial role in determining the physiological and pharmacological properties of the receptors.[68] Research involving GABAA receptor subtype-selective compounds and genetically engineered mice with subunit point mutations has provided valuable insights into the specific roles of different subunits in the clinical effects of BDZs. BDZs do not open the chloride channel in the absence of GABA.[69]Various pharmacological agents, including GABA, GABAA receptor agonists like muscimol, and antagonists like bicuculline, act on different sites of the GABAA receptor. Classic BDZs, such as chlordiazepoxide and diazepam, bind to the GABAA modulatory site, which is distinct from other sites where compounds like alcohol, barbiturates, and neurosteroids bind.[70] The allosteric binding site for BDZs is always formed by two ?-subunits (?1, 2, 3, or 5), two ?-subunits, and the ?2 subunit. BDZs act as allosteric modulators, altering the efficacy and/or affinity of GABA in a positive (positive allosteric modulation, PAM), negative (NAM), or neutral manner by stabilizing different three-dimensional conformations of the GABAA receptor complex. The specificity of a BDZ ligand for a particular receptor subtype, as well as its affinity and efficacy, determines its potency and clinical effects.[71] BDZs have a broad range of effects, including anxiolytic, sedative, hypnotic, muscle-relaxant, and anticonvulsant properties. However, when anxiolysis is the primary therapeutic goal, the sedative and hypnotic effects are often undesirable. Classic BDZs non-selectively activate ?1, 2, 3, and 5 subunits, which leads to these unwanted effects. Extensive research, particularly in genetically engineered mice where specific GABAA receptor ?-subunits were rendered insensitive to BDZ binding while preserving GABA binding, has shown that different subunits contribute to different functions. The wide range of therapeutic and side effects of BDZs can be attributed to the activation of various ?-subunits.[72]
Preclinical studies have implicated the ?2 and ?3 subunits in anxiety modulation. However, efforts to develop new, highly specific anxiolytics targeting these subunits have not yet been successful, with issues like tolerance and dependence still present in new ligands. The question remains whether activating all ?-subunits leads to tolerance and addiction or if targeting a specific subunit could avoid these issues. The development of Z-drugs, such as zolpidem, zaleplon, and zopiclone, initially showed promise but did not result in anxiolytics free from tolerance and dependence problems.[73] Interestingly, pregabalin, a structural analogue of GABA, has demonstrated anxiolytic effects through a mechanism distinct from that of BDZs and was approved for anxiety disorders in the EU in 2007, though not in the USA. Evidence suggests that the activation of ?1-subunits plays a critical role in the addictive potential of BDZ ligands, but the mechanisms underlying tolerance development are complex and depend on the specific endpoint being measured. The challenges associated with BDZs and the lack of progress in developing new anxiolytics targeting the GABAA receptor-BDZ complex have likely contributed to the pharmaceutical industry's retreat from this area of drug development.[74]
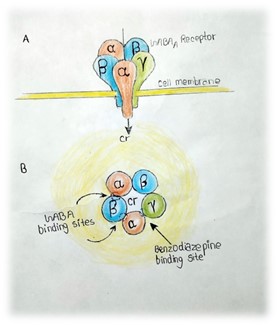
Fig. 6. This schematic illustration depicts the GABAA receptor. Part A presents an overview showing the arrangement of two ? subunits, two ? subunits, and one ? subunit, along with the direction of chloride ion influx through the receptor. Part B offers a top-down view, revealing the subunits organized around a central pore that functions as the chloride channel. The binding sites for GABA and benzodiazepines (BDZ) are also indicated in this view.
The Role of CRH and HPA Axis in Anxiety
Hyperactivity of the hypothalamic-pituitary-adrenal (HPA) axis is linked to increased synthesis and release of corticotropin-releasing hormone (CRH) from hypothalamic neurons in the paraventricular nucleus (PVN) in response to stress. CRH-expressing neurons have also been identified in other brain regions, such as the central amygdala (CeA), which can activate the HPA axis through projections to the PVN.[75] Additionally, reciprocal connections between these CRH neurons and aminergic nuclei, including the locus coeruleus (LC) and raphe nuclei (RN), facilitate interaction between the noradrenergic and serotonergic systems with the HPA axis during stress responses. CRH neuronal circuits interact with these systems, both of which play crucial roles in mood and anxiety disorders. Moreover, CRH is associated with anxiety and the encoding of emotional memories, underscoring its critical role in the stress response and its long-lasting impact, particularly following early life stress.[76]
The impact of traumatic events during childhood, such as abuse, neglect, or loss, is a critical factor in the development of mood and anxiety disorders later in life. These early adverse experiences are linked to alterations in various limbic structures and the HPA axis. Early stress exposure may result in decreased availability and reduced efficacy of hippocampal glucocorticoid receptors (GRs), leading to glucocorticoid resistance and heightened HPA axis reactivity in response to stress later in life.[77] Furthermore, elevated cortisol levels combined with decreased GR availability due to early stress are associated with diminished hippocampal function and reduced volume in adulthood. Consequently, early adverse events can cause enduring changes, including hyper-reactivity of neural and neuroendocrine responses to stress, increased CRH levels, glucocorticoid resistance, and reduced hippocampal volume, all of which may influence responses to future stress.[78]
HPA Axis Dysregulation in Anxiety and the Impact of Antidepressant Treatment
A significant number of individuals with chronic anxiety disorders display hyperactivity of the hypothalamic-pituitary-adrenal (HPA) axis, leading to elevated cortisol levels. This hypercortisolism has been observed in patients with panic disorder and generalized anxiety disorder (GAD). However, studies have shown mixed results regarding hypercortisolism in GAD, with some reporting it and others not.[79] In contrast, patients with post-traumatic stress disorder (PTSD) often exhibit decreased HPA axis activity, which has been linked to exaggerated negative feedback or hypersecretion of corticotropin-releasing hormone (CRH), resulting in downregulation of CRH receptors in the anterior pituitary.[80] Pharmacological treatments that successfully alleviate anxiety symptoms often normalize the HPA axis, suggesting a role for this system in the pathophysiology of anxiety disorders. Different classes of anti-anxiety medications, including tricyclic antidepressants (TCAs), selective serotonin reuptake inhibitors (SSRIs), and benzodiazepines (BZDs), have been shown in some studies to regulate HPA axis activity.[81] This has prompted research into the broader mechanisms of action of these agents, particularly their effects on the transcriptional regulation of molecules involved in HPA axis regulation, such as glucocorticoid receptors (GRs), mineralocorticoid receptors (MRs), and CRH.[82]
Studies have shown that TCAs and SSRIs not only block neurotransmitter reuptake but also induce significant changes in the HPA axis, which may contribute to their therapeutic effects. For example, altered GR gene regulation,[83] leading to reduced GR concentrations in neural structures like the hippocampus or hypothalamus, may result in impaired feedback regulation of the HPA axis. This dysregulation could contribute to the abnormalities observed in patients with depression or chronic anxiety disorders.[84] In vitro studies have demonstrated increased GR mRNA expression in hypothalamic or amygdala cell cultures treated with TCAs, such as desipramine and amitriptyline. Similar findings have been observed in vivo, where chronic TCA treatment, but not short-term treatment, reduced CRH mRNA expression in the hypothalamus, leading to decreased HPA axis activity.[85] Long-term TCA administration, such as imipramine, has also been shown to inhibit CRH gene transcription, further reducing CRH expression in the hypothalamus and decreasing HPA axis reactivity.[86] For SSRIs, both in vitro and in vivo studies have shown that long-term fluoxetine treatment increases GR mRNA expression in hippocampal neurons. Additionally, fluoxetine has been found to promote functional recovery of hippocampal GRs following chronic stress and to enhance GR activation, including phosphorylation and nuclear translocation, even without changes in glucocorticoid secretion. These findings suggest that GR modulation may play a role in the therapeutic effects of fluoxetine, though some studies indicate that changes in GRs are not always necessary for SSRI efficacy.[87] GRs are also expressed in the amygdala, particularly in the central nucleus (CeA), where glucocorticoids stimulate CRH expression, in contrast to the inhibitory effects observed in the hypothalamic paraventricular nucleus (PVN).[88] Cortisol-induced CRH upregulation in the amygdala may activate the entire HPA axis, as CRH projections from the CeA can stimulate the PVN, increasing CRH synthesis and release in the hypothalamus and contributing to HPA axis hyperactivity.[89] Hyperactivity of the amygdala is frequently associated with depression and chronic anxiety disorders, as demonstrated in functional imaging studies. The amygdala's role in anxiety pathophysiology, particularly through CRH projections from the CeA, is critical for HPA axis regulation.[90] The amygdala is a major source of extra-hypothalamic CRH, and its hyperactivation may lead to elevated CRH levels in cerebrospinal fluid (CSF), as seen in patients with depression and in animal models of chronic stress.[91] CRH overexpression in the CeA has been proposed as a key factor in the development of depression. Therefore, SSRIs that reduce GR and CRH gene expression in the CeA may help downregulate the HPA axis, contributing to clinical improvement.[92] Studies on escitalopram, an SSRI, have shown that it can normalize various HPA axis-related physiological parameters. Escitalopram has been effective in reducing elevated cortisol levels in GAD patients, correlating with clinical improvement.[93] In vivo studies have also demonstrated that escitalopram reduces CRH expression in the hippocampus while increasing GR expression in the hypothalamus and hippocampus, leading to decreased HPA axis reactivity.[94] Additionally, preclinical studies have shown that escitalopram inhibits CRH and its receptor expression in the hypothalamus.[95]
Understanding the potential effects of TCAs and SSRIs on HPA axis regulation may provide further insights into their therapeutic mechanisms, although more research is needed to explore this critical issue fully.[96]
Molecular Mechanisms Underlying the Long-Lasting Effects of TCAs and SSRIs: Involvement of the AC-cAMP-PKA Cascade
To elucidate the molecular mechanisms responsible for the prolonged effects of tricyclic antidepressants (TCAs) and selective serotonin reuptake inhibitors (SSRIs), various studies have explored their impact on different components of the hypothalamic-pituitary-adrenal (HPA) axis[97] It has been proposed that these long-lasting effects may involve the upregulation of the cAMP-mediated second messenger cascade, which could lead to the transcriptional regulation of various genes, including glucocorticoid receptor (GR), corticotropin-releasing hormone (CRH), and brain-derived neurotrophic factor (BDNF).[98] Activation of G protein-coupled receptors (GPCRs) by various ligands triggers second messenger cascades. Specifically, stimulation of Gs-coupled receptors can activate adenylate cyclase (AC), leading to the synthesis of cAMP, which in turn activates cAMP-dependent protein kinase (PKA).[99] This cascade ultimately results in the activation of cAMP response element-binding protein (CREB), a transcription factor that mediates the effects of the cAMP pathway.[100] CREB becomes active upon phosphorylation at a specific serine residue (Ser133), allowing it to regulate gene transcription by binding to cAMP response elements (CREs) in the promoter regions of target genes.[101] The AC-cAMP-PKA-CREB pathway can also be activated via calcium-dependent protein kinase (PKC) cascades, making CREB a converging point for various stimuli, including those mediated by TCAs and SSRIs.[102] Alterations in this cascade have been observed in patients with depression, and chronic antidepressant treatment has been shown to restore this pathway at different molecular levels. This restoration supports the critical role of CREB in regulating target genes, such as CRH, at the transcriptional level.[103] In vivo studies have demonstrated that chronic treatment with TCAs or SSRIs increases CREB expression in specific limbic regions, particularly in the hippocampus.[104] These studies have shown elevated CREB mRNA levels in hippocampal regions such as CA1, CA3 pyramidal cells, and dentate gyrus granule cells following prolonged antidepressant .[105] Additionally, increased BDNF mRNA expression in the hippocampus has been observed, suggesting that chronic antidepressant treatment may upregulate CREB, which in turn enhances the expression of genes like BDNF that contain CREs in their promoter regions.[106] The upregulation of CREB and the resulting increase in BDNF expression may be crucial for counteracting the effects of stress on hippocampal neurons.[107] BDNF also plays a role in regulating the HPA axis, providing a potential link between the AC-cAMP-PKA cascade, CREB activation, and HPA axis regulation. However, the exact molecular mechanisms involved remain unclear and require further investigation.[108] CREB is associated with neuronal survival and plasticity in the hippocampus, and its increased expression has been linked to the therapeutic effects of antidepressants, suggesting that CREB could be a potential target for the development of new therapeutic agents.[109] The therapeutic effects of TCAs and SSRIs have also been linked to the upregulation of 5-HT1A heteroreceptors, which primarily function through Gi proteins, thereby inhibiting the AC-cAMP-PKA signaling cascade. The paradox of TCAs and SSRIs activating the AC-cAMP-PKA pathway while 5-HT1A receptors inhibit it may be explained by the indirect signaling mechanisms of 5-HT1A receptors.[110] Activation of hippocampal 5-HT1A receptors, particularly in the dentate gyrus, has been associated with the inhibition of GABAergic interneurons. Additionally, other GPCRs, such as 5-HT4 receptors, which are coupled to Gs proteins, have been found to stimulate the AC-cAMP-PKA cascade, leading to CREB phosphorylation and increased BDNF synthesis.[111] The activation of 5-HT4 receptors has also been associated with the therapeutic effects of SSRIs, suggesting interactions between 5-HT1A and 5-HT4 receptors in the antidepressant mechanism of action.[112] Furthermore, 5-HT1A receptors can form heterodimers with other GPCRs, such as 5-HT7 receptors. The 5-HT7 receptor, coupled to Gs, stimulates the AC-cAMP-PKA cascade upon activation.[113] Heterodimerization of 5-HT1A and 5-HT7 receptors has been shown to modulate downstream signaling pathways, potentially altering their regulatory effects. This interaction represents a complex molecular mechanism that could contribute to the therapeutic effects of antidepressants.[114]
GABAergic Regulation of the HPA Axis and Its Implications for Anxiolytic Therapy
The hypothalamic-pituitary-adrenal (HPA) axis, a critical component of the body's stress response, is influenced by various neurotransmitters, including ?-aminobutyric acid (GABA), the primary inhibitory neurotransmitter in the central nervous system (CNS). GABA plays a significant role in regulating hypothalamic function, particularly by modulating corticotropin-releasing hormone (CRH) neurons located in the medial parvocellular region of the paraventricular nucleus (PVN) of the hypothalamus.[115] GABAergic input to these CRH neurons can be direct, originating from peri-PVN sources such as adjacent hypothalamic nuclei and the bed nucleus of the stria terminalis (BNST), or indirect, arriving via cortical and limbic structures like the hippocampus, amygdala, and prefrontal cortex (PFC), including the anterior cingulate cortex (ACC), prelimbic, and infralimbic areas. These pathways are integral to the adaptive regulation of the HPA axis in response to stress.[116]At the molecular level, GABA exerts its effects through two types of postsynaptic receptors: GABAA and GABAB. The GABAA receptor, a complex structure composed of various subunits, contains specific binding sites for its natural ligand GABA, as well as for benzodiazepines (BZDs) and barbiturates. Upon GABA binding, GABAA receptors open chloride ion channels, leading to chloride influx, membrane hyperpolarization, and subsequent inhibition of neuronal firing. Benzodiazepines enhance GABA's binding to GABAA receptors, increasing the frequency of chloride channel opening and thus amplifying the inhibitory effects on neurons.[117,118] This mechanism underpins the anxiolytic properties of benzodiazepines, which have been shown to modulate the HPA axis, particularly in individuals with anxiety and depression. For instance, alprazolam, a potent benzodiazepine agonist, has been demonstrated to exert an inhibitory effect on the HPA axis, reducing CRH activity in the hypothalamus. Similarly, diazepam has been found to lower corticosterone levels in animal models and decrease cortisol levels in both healthy volunteers and depressed patients, with these effects occurring in a dose-dependent manner.[119] Further studies have shown that the therapeutic efficacy of benzodiazepines may be partly attributed to their influence on CRH neurons within the hypothalamus and the locus coeruleus (LC), a brain region rich in CRH innervation and critically involved in the stress and anxiety response. Both acute and chronic administration of alprazolam has been shown to reduce CRH concentrations in the LC, suggesting that the drug's anxiolytic effects may involve both direct and indirect modulation of CRH activity.[120] In addition, recent in vivo studies with lorazepam and clonazepam have confirmed their ability to reverse anxiety-like behaviors, including social avoidance, which correlated with their suppression of the HPA axis through CRH inhibition. These findings underscore the significance of GABAergic modulation of the HPA axis in the pharmacological treatment of anxiety disorders.[121] Despite these insights, the effects of anxiolytics, including benzodiazepines, tricyclic antidepressants (TCAs), and selective serotonin reuptake inhibitors (SSRIs), on the HPA axis represent only a portion of their therapeutic actions. While these mechanisms contribute to our understanding of their pharmacological effects, they account for only a small part of the overall improvement seen in hypercortisolism and other symptoms in patients with depression or anxiety disorders.[122]
Therapeutic Challenges in Anxiety Disorders
The pursuit of novel pharmacotherapies for anxiety disorders has encountered substantial obstacles despite extensive preclinical exploration. While the GABAergic and serotonergic systems have historically been the primary focus, the limitations of these targets have necessitated a broadening of research to encompass a wider range of molecular mechanisms.[123]
Neuropeptide systems, including those involving CRF1, CCK2, and neurokinin1/2 receptors, have garnered attention due to their established roles in stress and anxiety responses. Preclinical data consistently support the anxiolytic potential of antagonists targeting these receptors. However, the transition from bench to bedside has been fraught with challenges, underscoring the complex nature of anxiety disorders and the limitations of preclinical models in predicting clinical outcomes.[124] The glutamatergic system, characterized by its intricate architecture and diverse receptor subtypes, presents both opportunities and challenges for drug discovery. While preclinical studies have implicated glutamate dysregulation in anxiety, the development of selective and efficacious modulators has proven elusive. Metabotropic glutamate receptors (mGluRs) have been extensively investigated, but clinical trials have yielded disappointing results.[125] A paradigm shift in the treatment of anxiety disorders emerged with the discovery of ketamine's rapid and robust anxiolytic effects in treatment-resistant populations. Ketamine's unique mechanism of action, targeting the NMDA receptor, offers a promising alternative to traditional approaches. Nevertheless, the challenges associated with its administration, safety profile, and abuse potential necessitate the development of safer and more clinically viable derivatives.[126] The complexities inherent in translating preclinical findings to effective clinical treatments highlight the need for a more comprehensive understanding of the neurobiological underpinnings of anxiety disorders. Moreover, innovative approaches to drug discovery, including the development of novel target identification strategies and refined preclinical models, are essential to address the unmet medical needs of patients with anxiety.[127] Ultimately, overcoming the challenges associated with anxiety drug development will require a multidisciplinary approach that integrates basic neuroscience, pharmacology, and clinical research. By fostering collaboration and knowledge sharing among these disciplines, the field can accelerate the discovery and development of novel and effective treatments for anxiety disorders.[128]
CONCLUSION
The complex nature of anxiety disorders presents significant challenges in developing effective pharmacological treatments. Despite decades of research focusing on key neurotransmitter systems like GABA and serotonin, as well as the HPA axis, a comprehensive understanding of the underlying neurobiological mechanisms remains elusive. The limitations of current therapies, including SSRIs and benzodiazepines, highlight the need for innovative approaches to drug discovery and development. Emerging research into neuropeptide systems, glutamatergic modulation, and rapid-acting agents like ketamine offers promising new avenues for investigation. However, the difficulties in translating preclinical findings to clinical success underscore the importance of refining animal models and developing more predictive screening methods. Moving forward, a multidisciplinary approach that integrates insights from neuroscience, pharmacology, and clinical research will be crucial. This may involve exploring novel molecular targets, investigating gene-environment interactions, and leveraging advanced technologies like optogenetics and CRISPR/Cas9 to elucidate anxiety mechanisms at a cellular level. Additionally, recognizing the heterogeneity of anxiety disorders and embracing personalized medicine approaches could lead to more targeted and effective treatments. By considering individual genetic profiles, environmental factors, and specific symptom clusters, clinicians may be able to tailor interventions more precisely. Ultimately, overcoming the therapeutic challenges in anxiety disorders will require sustained collaborative efforts across scientific disciplines. As our understanding of the intricate neural circuits and molecular pathways underlying anxiety continues to evolve, so too will our ability to develop innovative, safe, and efficacious treatments that significantly improve the lives of those affected by these pervasive and debilitating conditions.
REFERENCES


















K. Rajeswar Dutt , Maheswar Prasad Deep , Abrarul Haque , Archana Rani , Nahida Khatun , Ayush Kumar , Subham Kumar Lohani , Sajid Ansari , Balraj Kumar , Nisha Kumari , Pratik Mondal , Sudarshan Rawani , Gangadhar Singh , Suraj Kumar , Rashmi Kumari , Arnab Roy , Unraveling The Neurobiological Mechanisms And Therapeutic Challenges In Anxiety Disorders: A Comprehensive Review, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 9, 458-483. https://doi.org/10.5281/zenodo.13732139
 10.5281/zenodo.13732139
10.5281/zenodo.13732139