
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmacy Practice, Vaageswari College of Pharmacy, Karimnagar, Telangana, India
Pharmacovigilance (PV) serves as the cornerstone of global healthcare, evolving from a reactive monitoring system into a proactive, multidisciplinary science dedicated to advancing patient safety.The fundamental objective of pharmacovigilance is the continuous assessment of the risk-benefit balance of pharmaceutical products throughout their entire lifecycle. A significant focus is placed on spontaneous reporting systems, which remain the primary source of safety data.Regulatory authorities, including the FDA (USA), EMA (Europe), and CDSCO (India), provide the essential legal and ethical scaffolding for these activities. They enforce compliance with Good Pharmacovigilance Practices (GVP) and possess the mandate to take corrective actions ranging from updating safety labels (Black Box Warnings) to the complete withdrawal of products from the market. In 2026, global harmonization between these agencies is more critical than ever to manage the safety of complex biologics, biosimilars, and personalized therapies across international borders. Advancing patient safety necessitates a seamless synergy between technological innovation in PV processes, a robust culture of ADR reporting, and stringent regulatory enforcement. By fostering transparency and global collaboration, the pharmacovigilance ecosystem continues to minimize medicinal risks and optimize public health outcomes.
The safety profile of a pharmacological product is a dynamic continuum that extends far beyond the controlled environments of clinical trials. While pre-marketing clinical studies provide essential baseline information on efficacy and common adverse events, they are fundamentally limited in their ability to capture unexpected, delayed, or long-term toxicities that manifest only in diverse, real-world patient populations. This fundamental limitation necessitates pharmacovigilance, a discipline that serves as the critical sentinel for public health. The WHO defines Pharmacovigilance (PV) as “the science and activities related to the detection, assessment, understanding, and prevention of adverse drug reactions or any other possible drug-related problems”. It is the fundamental component of effective drug regulation systems, public health programmes, and clinical practice. An Adverse event is defined as “any untoward medical occurrence that may present during treatment with a drug but does not necessarily have a relationship with its use”. An Adverse drug reaction is “any noxious, unintended, and undesired effect of a drug that occurs at a dose used in humans for prophylaxis, diagnosis, therapy, or modification of physiological function” [1].
Pharmacovigilance (PV) was officially introduced in December 1961 with the publication of a letter (case report) in the Lancet by W. McBride, the Australian doctor who first suspected a causal link between serious fetal deformities (phocomelia) and thalidomide, a drug used during pregnancy: Thalidomide was used as an antiemetic and sedative agent in pregnant women [2]. In 1968, the World Health Organization (WHO) promoted the “Programme for International Drug Monitoring”, a pilot project aimed at centralizing world data on adverse drug reactions (ADRs). In particular, the main aim of the “WHO Programme” was to identify the earliest possible PV signals. The term PV was proposed in the mid-70s by a French group of pharmacologists and toxicologists to define the activities promoting “The assessment of the risks of side effects potentially associated with drug treatment.”
PV aims to reduce medication-related risks and protect public health by detecting and identifying adverse drug reactions (ADRs), at-risk populations, and product safety [3]. The development of drugs occurs within a small, selected population. However, pharmacovigilance assesses the safety of drugs in real-world scenarios, which include a diverse population and prolonged drug use [4].
Promoting pharmacovigilance techniques is extremely difficult in low- and middleincome countries (LMICs ) due to factors like low awareness, low reporting rates, widespread use of traditional medicines, a lack of qu alified staff, and restricted access to drug usage data [5]. In clinical trials, pharmacovigilance plays a major role in suspecting ADRs in study participants through clinical examination, lab reports, and submitting information to regulatory authorities during drug approval [6]. Databases such as VigiFlow, VigiAccess, VigiBase, and VigiLyze are used by the Uppsala Monitoring Center, along with Vigi Grade, Vigi Match,and VigiRank for case report analysis [7]. Advanced technologies that optimise the drug's benefitrisk profile in practical situations include electronic data capturing and machine learning (ML) techniques [8]. The pharmacovigilance process consists of four stages: detection, assessment, understanding, and prevention of adverse drug reactions [9]. The Pharmacovigilance Programme of India (PvPI), with 250 monitoring centers around India, became a WHO collaborating center for pharmacovigilance. This comprehensive review gives a brief explanation of the history of Pharmacovigilance in India, types of adverse drug reactions, signal detection, clinical trials, risk-benefit analysis, post-marketing surveillance, Materiovigilance, software used in PV, and the regulatory framework of pharmacovigilance.
HISTORY :
In 1848, a young England girl named Hannah Grenner died from pulmonary aspiration and arrhythmia after the use of chloroform as an anaesthetic to remove an infected toenail [10]. Likewise, many episodes occurred in 1937, when Sulfanilamide (Prontosil), already in use since 1932 for treatment of streptococcal infections, was launched as a syrup, containing Diethyleneglycol as a solvent.The drug was responsible for the death of 105 individuals (34 children and 71 adults) and Diethyleneglycol was incriminated. This tragedy caused the American Congress to approve the Food Drug and Cosmetic Act, in 1938 [11]. The 1961 Thalidomide tragedy changed European pharmacovigilance, resulting in the development of the E C Directive 65/65 in the USA and the "Yellow card" in the UK [12]. In India, clinical trials were globally recognized in the year 1996. In 1997, India joined with the WHO Adverse Drug Reaction Monitoring Program and it was not successful. India established its 67th National Pharmacovigilance Center in 2002.The Pharmacovigilance Program of In dia (PvPI) took the responsibilities of India's National Pharmacovigilance Program, which was started in 200 5 [13]. The mission of PvPI is to safeguard the health of the Indian population by ensuring that the benefits of use of medicine outweigh the risks associated with its use.
The Central Drug Standard Control Organization (CDSCO) established the PvPl program, but later the responsibilities of the program were taken by the Indian Pharmacopeia Commission in Ghaziabad on 15 April 2011. The primary aim of the PvPI is to safeguard the health of the Indian population by preventing and reporting ADRs. PvPI coordinates with 250 adverse drug monitoring centers within India, and it became a WHO Collaborating Centre for Pharmacovigilance [12,13].
AIMS AND OBJECTIVES OF PvPI :
17th September to 23rd September every year
PHARMACOVIGILANCE CENTERS IN INDIA :
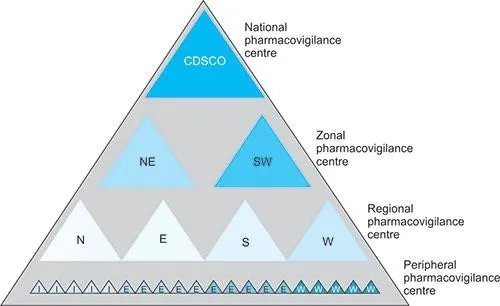
Figure 1: Pharmacovigilance centers in India
ADR Monitoring Centres (AMC’s) are responsible for the collection and reporting of ADRs to PvPI NCC, answering queries and checking for follow-ups, entry of collected data into databases such as vigiflow, and providing feedback to physicians.
PvPI-NCC (PvPI National Coordinating Centre): helps in developing standard operating procedures (SOPs), casuality assessment,reporting of ADRs to CDSCO, and analysis of cases.
Zonal or Sub-zonal centers are helpful for financial and managerial help to AMC (Zonal centers are located in Ahmedabad, Hyderabad, Ghaziabad, Kolkata, Mumbai, and Chennai).
CDSCO, New Delhi : It takes the major decisions, provides recommendations, and collaborates with WHO- UMC.
LIST OF CENTRAL DRUG STANDARD CONTROL ORGANISATION (CDSCO) ZONAL AND SUB-ZONAL OFFICES :
Zonal Centre: Ahmadabad
Zonal Centre: Hyderabad
North Zonal centre: Ghaziabad a) Sub-Zone Office-Ghaziabad, b) Chandigarh, c) Sub-Zonal Office, Jammu.
East Zonal Centre: Kolkata-Air Port and Sea Office, Kolkata.
West Zonal Centre: Mumbai-Air Port and Sea Office, Mumbai, Jawaharlal Nehru Port Office, Navi Mumbai
South Zonal Centre: Chennai, a) Airport and Sea Office, b) Sub-Zonal and Port Office, Chennai; c) Port Office, Kochi, Bangalore [15] as shown in Figure 1.
PROCESS OF PHARMACOVIGILANCE: As shown in Figure 2
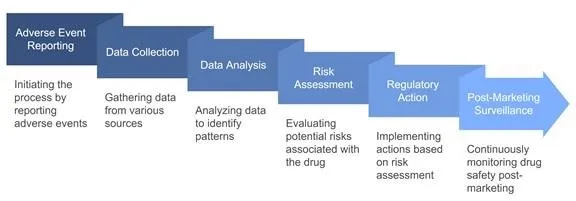
Figure 2 : Steps in the pharmacovigilance program
A.SIGNAL DETECTION :
Signal detection in pharmacovigilance refers to the identification of newly discovered drug-drug interactions, new unlabeled adverse events, or increased frequency, severity, or specificity of a labeled event. In addition to identifying a drug’s adverse effects, signal detection can also identify new populations that may be at danger or new beneficial impacts.Reporting the causal connection between a medication and an adverse event is beneficial [14,15]. Among the sources of signal detection are:
I)Types of ADRs :
a)Type A (Augmented reactions) is strongly related to the medication's dosage. Hepatic disorders, genetic polymorphisms, electrolyte imbalances, etc., could possibly increase the effects of drugs. Clotting factors may be produced as a result of hepatic disorders. Pharmacodynamics changes can also be exacerbated by hypokalaemia and hypercalcemia brought on by cardiac glycoside. Due to their limited specificity, ADRs must be quantitatively and carefully determined. This will support the connection and persistence of these adverse reactions.[16 ,17]
b)Type B (Bizarre reactions ) ADRs have little or no relation with dosage and are sensitive and unpredictable. ADRs of type B are divided into two categories: immunological and non-immunological. Overdose, collective effects, delayed toxicity, drugs interactions, metabolic changes, and other factors make non-immunological reactions predictable adverse drug reactions (ADRs). Unpredictable immunological Type B reactions might result from immunological reactions or from cell-mediated, cytotoxic drug-induced reactions. The unusual absorption and distribution could be the pharmacokinetic cause of type B adverse drug reactions. The individual’s vulnerability to the particular medication determines the pharmacodynamics reactions of type B adverse drug reactions.[16,17]
An ICSR is a crucial component of pharmacovigilance because it provides comprehensive information on the type of adverse drug reaction, which helps pharmaceutical companies and regulatory organizations monitor safety as well as minimize risk. There are various ways to submit an individual case report, including spontaneous reporting, reporting by using existing literature, regulatory authority reports, reports from clinical trials, post-marketing surveillance, and solicited reports.
Spontaneous reports are gathered from volunteers and submitted by healthcare experts, pharmaceutical companies, or patients themselves. Certain reports may lack sufficient clinical data yet are utilized for trend analysis and signal detection.
Published literature, including case series, case reports, and systematic reviews, functions as a source for reporting, and this reporting system is termed a literature report. They are reported by pharmaceutical companies to the regulatory authorities after rigorously checking the databases and medical publications.
Based on collection of data from clinical studies and spontaneous reports, regulatory bodies report to regulatory authorities. This kind of reporting includes aggregated data on drug safety and is helpful for regulatory organizations' decision-making, which eventually helps in updating labels and giving safety warnings are regulatory authority reporting.
Clinical trial reporting systems, which follow Good Clinical Practice (GCP) principles, are used to report adverse drug reactions (ADRs) during clinical trial phases. These adverse drug reactions should often be reported within 7 to 15 days. It includes complete information regarding the patient’s health, medical history, causality assessment, etc. Serious adverse events and unexpected serious adverse events make up the majority of ADRs recorded by the clinical trial reporting system. This kind of reporting system is helpful for regulatory authorities to approve the medication.
One kind of ADR reporting system that gathers information from several sources, including patient registries,
PMS studies, and risk management initiatives to support risk reduction tactics, is the Post Marketing Surveillance (PMS) Report. It aids in improving a drug’s safety profile by identifying any rare or delayed ADRs from large amounts of data.
A Solicited Reporting system evaluates actual medication safety by using survey data collected using predetermined proforma or systematic forms.
Legal Case Reports Adverse events identified through product liability claims or litigation. These often involve serious outcomes and require careful handling due to legal implications and confidentiality considerations.
License Partner Reports Safety information exchanged between partners in licensing agreements (e.g., comarketing, distribution partnerships). Requires clear data exchange agreements and responsibility matrices to avoid duplicate reporting.
Vaccine Adverse Event Reporting System (VAERS)
The Food and Drug Administration (FDA) and the Centers for Disease Control and Prevention (CDC) monitor vaccine safety issues through the Vaccine Adverse Event Reporting System (VAERS). All the adverse event reports are collected and reviewed by VAERS to promote safety and it determines whether further investigations are needed or not.CDC encourages reporting adverse events by anyone who experiences adverse events after receiving the vaccine. Clinically significant adverse events should be reported to the FDA to promote the safety of vaccines [18]. In India, adverse events are monitored by the Adverse Event Immunization Surveillance System.
III) Regulatory authorities in Pharmacovigilance:
The common goal of safeguarding public health is shared by three major regulatory bodies: the Central Drug
Standard Control Organisation (CDSCO) of India, the European Medicines Agency (EMA), and the United States Food and Drug Administration (US FDA).
a)United States Food and Drug Administration (US FDA) : The US FDA is the primary regulatory body in the United States and is responsible for overseeing food, drugs, biologics, medical devices, and cosmetics.
Established in 1906, the FDA operates under the Department of Health and Human Services.
Approval Process :
b)European Medicines Agency (EMA) : The EMA is the regulatory body responsible for the evaluation and supervision of medicinal products in the European Union (EU). Established in 1995, it collaborates with national agencies of EU member states.
Approval Process :
c)Central Drugs Standard Control Organization (CDSCO): The CDSCO is India’s national regulatory authority, operating under the Ministry of Health and Family Welfare. It regulates pharmaceuticals, biologics, and medical devices in India. The Ministry of Health and Family operates India’s major regulatory authority, the CDSCO. It is responsible for receiving applications of Investigational New Drug (IND), approval to conduct clinical trials, New Drug Approval, and Subject Expert Committee (SEC) Review. The organization adheres to the Drugs and Cosmetics Act, 1940 and Rules, 1945, collaborates with state regulatory authorities, and expedites approvals for critical drugs during emergencies.[19]
B. CASE PROCESSING AND MANAGEMENT :
Case processing is crucial for safety monitoring in pharmacovigilance. The process of determining the probability that a particular medication, vaccination, or other medical intervention would result in an adverse event is known as a causal assessment[20]
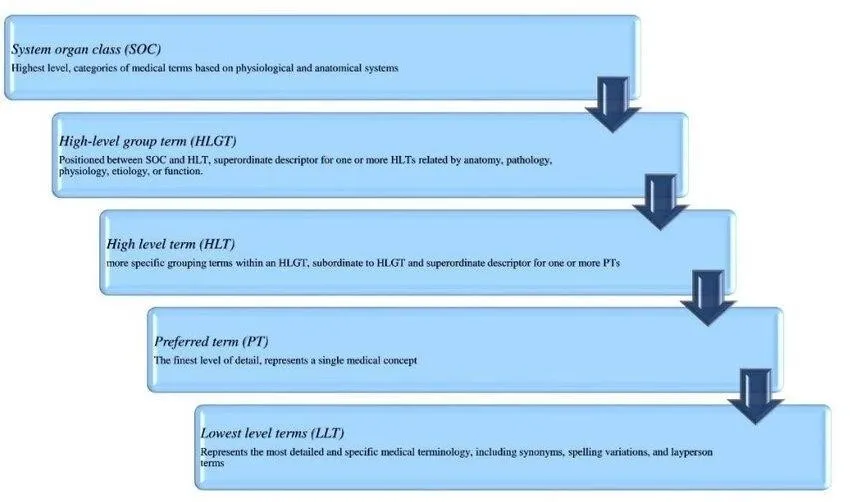
Figure 3 : Med DRA five-level structure:Medical Dictionary for Regulatory Activities (MedDRA) Overview
C. EVALUATION: DATA MINING AND STATISTICAL ANALYSIS :
Data mining in pharmacovigilance involves the extraction of meaningful patterns from large datasets. These patterns help in detecting signals—an indication that a drug may be associated with an ADR. Various techniques are employed in data mining to uncover these signals, each with its strengths and specific applications. Disproportionality Analysis is one of the most common techniques used in pharmacovigilance.Key methods include Proportional Reporting Ratio,Reporting Odds Ratio,Bayesian
Confidence Propagation Neural Network (BCPNN), Bayesian Confidence Propagation Neural Network
(BCPNN).Empirica Signal, developed by Oracle Health Sciences, is a leading software platform designed to support pharmacovigilance professionals in detecting and analyzing drug safety signals. Statistical techniques are used for data mining that are similar to conventional methods of examining data, but there is no prior hypothesis, and power calculations are performed.
D.RISK ASSESSMENT AND RISK BENEFIT EVALUATION
Benefit-risk assessment is an important component of Pharmacovigilance: it is used to support regulatory and clinical decision making affecting vast populations of patients.Initially, the benefits and risks of the product should be identified for conducting a risk-benefit analysis. The second step is the collection of the data, where information from clinical trials, preclinical studies, and post-marketing surveillance is gathered to assess the safety and efficacy of a drug product [23]. Several techniques are employed for risk-benefit analysis, which include Benefit Less Risk Analysis (BLRA), Risk Benefit Assessment for
Drug Safety, Quality Adjusted Time Without Symptoms and Toxicity (Q-TWiST), Incremental Net Health
Benefit (INHB), Risk Benefit Plane (RBP) & Risk Benefit Acceptability Threshold (RBAT), Risk Benefit Contour (RBC), and Minimum Clinical Efficacy (MCE)[24].
E.SUBMISSION TO REGULATORY AGENCIES :
The submission process to regulatory agencies in pharmacovigilance involves capturing adverse event data, assessing it for causality and severity, and reporting it electronically (e.g., ICSRs, PSURs) within strict timelines.The Periodic Safety Update Report (PSUR) is helpful for post-authorization surveillance, which evaluates the drug’s safety following approval. It is used to measure the advantages and disadvantages of a medication at a certain moment. It provides a thorough and accurate presentation of the data for a critical evaluation of a drug’s benefits over its risks. In India, the New Drugs and Clinical Trial Rules 2019 established that applicants submit Periodic Safety Update Reports (PSURs) detailing safety variations, post-authorization status, and safety variation. All the information about dosage forms, formulations, indications of a new drug, and its effects in a specific population when used should be submitted to regulatory authorities through PSUR. PSUR should be submitted every six months for the first two years and once yearly for the subsequent two years. In certain circumstances, regulatory authorities may extend the period to continue the submission. For certain adverse reactions, they should be submitted within 30 calendar days after the reporting, while serious adverse events should be reported within 15 days after the reporting [25].Some of the regulatory agencies shown in Figure 4.
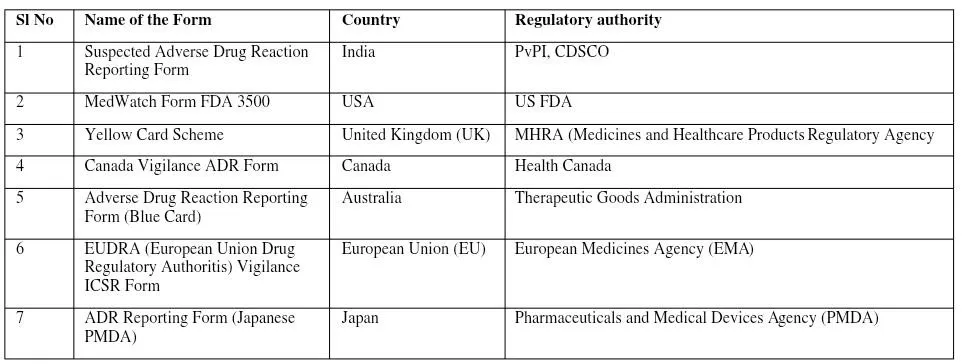
Figure 4 : Regulatory agencies of different countries and their means of reporting
F.RISK MANAGEMENT:
Risk management in pharmacovigilance refers to the systematic process of identifying, characterizing, preventing, or minimizing risks related to the use of pharmaceutical products.It spans both the clinical development and post-marketing phases and ensures that the benefits of a product outweigh its risks. Two major regulatory frameworks govern pharmacovigilance risk management globally are EU Risk Management Plan(RMP) and US Risk Evaluation and Mitigation Strategies (REMS). While the structures of RMPs and REMS differ, their shared goal is to ensure patient safety through targeted risk minimization and effective communication. Marketing Authorization Holders (MAHs) to deal with "Important identified risks", "Important potential risks", and "Important missing information" during approval review and post-marketing.
E.POST MARKETING SURVEILLANCE
While post-marketing surveillance is a real-world situation where a large number of people take the drug, clinical trials are conducted with a small number of participants. Pharmacovigilance is essential to postmarketing surveillance because it provides real-time data gathering and signal detection for evaluating safety concerns. Pharmacovigilance during post-marketing surveillance includes adverse event monitoring, signal detection, risk assessment, and regulatory compliance. Reports from patients, medical professionals, clinical research, and spontaneous reporting are all reviewed to keep monitoring on adverse events. The technique of finding possible adverse medication events using large databases from a variety of sources, including electronic health records, spontaneous reporting, and clinical trials, is known as signal detection. Risk assessment determines safety signals and severity of serious events. Depending on the country, pharmacovigilance complies with FDA, EMA, and CDSCO regulations.
CONCLUSION
The essential foundation of healthcare is pharmacovigilance (PV), which is the continuous surveillance system that ensures medications do more benefit than harm once they become available to everyone. The real-world reporting of adverse drug reactions (ADRs) is what enables regulatory agencies to detect unusual risks and update safety guidelines in real-time, even when clinical studies provide preliminary data. PV methods convert raw data into appropriate safety measures by bridging the gap between clinical observation and legal oversight. The integration of digital reporting technologies and international regulatory collaboration will be crucial as we move toward more advanced treatments. As medical science evolves, maintaining a strong, transparent system for tracking drug safety remains the most effective way to prevent harm, safeguard community health, and ensure that the benefits of modern medicine far outweigh the risks.
REFERNECES
https://www.clinskillacademy.com/data-mining-techniques-in-pharmacovigilance. Accessed on: 30 Mar 2025.
https://www.clinskillacademy.com/data-mining-techniques-in-pharmacovigilance. Accessed on: 30 Mar 2025.





Vaishnavi Shrinivas,Kappala SaiShivani, Veeragoni Jayadev, Pullela Akarsh, Sheelam Rajitha, Advancing patient safety: Pharmacovigilance processes, ADR reporting and Regulatory Authorities , Int. J. of Pharm. Sci., 2026, Vol 4, Issue 5, 6805-6816, https://doi.org/10.5281/zenodo.20392324
 10.5281/zenodo.20392324
10.5281/zenodo.20392324