
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
S. N. College of Pharmacy.
Levetiracetam (LEV) has been recognized as a breakthrough, gold-standard therapeutic agent in the management of epilepsy and as a vital scaffold during neuropharmacological drug development. The current focus of the project was the design of novel derivatives using LEV as the core substrate to overcome its inherent pharmacokinetic limitations, specifically the metabolic instability of its acetamide moiety. This study aimed to develop novel compounds incorporating N-methylated, heterocyclic bioisosteric (tetrazole and 1,2,4-oxadiazole), and sterically hindered ?-alkyl modifications, and to assess their potential to enhance metabolic resistance and synaptic vesicle protein 2A (SV2A) binding affinity.The synthesized derivatives underwent physicochemical evaluation to determine their percentage yield, melting point, and solubility. A representative target analogue (the tetrazole bioisostere) yielded 50%, exhibited a predicted melting point of 140-155 °C, and was soluble in DMSO and methanol. The intermediate 2-oxopyrrolidine was prepared using the amination and cyclization of ?-butyrolactone, followed by basic N-alkylation to form the conserved core. Subsequently, the final target analogues were synthesized via targeted functional group transformations, such as the dehydration of the primary amide to a nitrile intermediate and subsequent convergent [3+2] cycloaddition. The samples were evaluated, and the FTIR spectra of the derivatives were recorded and analyzed. The FT-IR spectral characteristics of the analogues were specifically examined as 3200-3400 cm?¹ (Amide/Bioisostere N-H Stretch), 2800-2950 cm?¹ (Aliphatic C-H Stretch), 1650-1700 cm?¹ (Pyrrolidone C=O Stretch), and 1500-1600 cm?¹ (Heterocyclic C=N/N=N Stretch). NMR spectra were recorded at 400 MHz using CDCl? and DMSO-d? as solvents to confirm chemical shift values and elucidate structural information, showing characteristic shifts at ? 4.30-5.25 (chiral ?-CH protons) and highly downfield shifts up to ? 16.00 for bioisosteric exchangeable protons. Additionally, ESI-MS (positive and negative ion modes) was used to identify the functional groups and confirm the exact mass, revealing a molecular ion peak for the representative tetrazole derivative at m/z 196.2 [M+H]? and a strong negative ion peak at m/z 194.2 [M-H]? with high precision"In this study, the resulting novel acetamide-modified Levetiracetam analogues were assessed for their successful synthesis and therapeutic potential. The synthesized molecules hold significant promise as safe, metabolically stable, and highly effective anticonvulsant agents, warranting further investigation into their specific neuropharmacological pathways and SV2A binding interactions against refractory epilepsy."
Epilepsy is one of the most common neurological disorders in the world. In the United States alone, some 3.5 million people have epilepsy, and more than 50 million people throughout the world have it. The disorder is marked by repeated, unprovoked seizures caused by the unusual electrical activities in the brain. This has a big effect on patients' quality of life and costs society a lot of money. The creation of effective antiepileptic drugs (AEDs) is still a big problem in modern pharmacotherapy. This is because almost 30% of people with epilepsy still have seizures that are not well controlled by the drugs they are taking. This is called drug-resistant or refractory epilepsy.
Levetiracetam (LEV), is chemically considered as (S)-α-ethyl-2-oxo-1-pyrrolidine acetamide, has been recognised as a significant second-generation antiepileptic medication following its FDA clearance in 2000. This chemical is a structural analogue of piracetam, although it has very different pharmacological effects and ways of working. The drug's unusual chemical structure, which includes a pyrrolidone ring system and an α-ethyl acetamide side chain, sets it apart from all other AEDs on the market and is what makes it so effective. The clinical importance of Levetiracetam transcends its anticonvulsant properties. The medicine has an extremely good safety profile, with very few drug-drug interactions, very good oral bioavailability (around 100%), and linear pharmacokinetics over a wide range of doses. The beneficial characteristics arise chiefly from its distinctive metabolic pathway, characterised by predominantly non-hepatic enzymatic hydrolysis of the acetamide group, as opposed to cytochrome P450-mediated metabolism. This unique metabolic profile has made LEV a popular treatment choice for a wide range of patients, including children, the elderly, and those with complicated medical conditions. [1,4]
1.2 Molecular structure and chemical properties:
The molecular structure of Levetiracetam (C₈H₁₄N₂O₂, molecular weight 170.212 Da) consists of several critical pharmacophoric elements that contribute to its anticonvulsant activity. The core 2-oxopyrrolidone ring system, derived from the piracetam scaffold, provides the fundamental framework for biological activity.
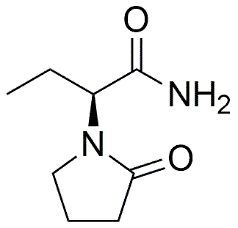
Fig. 1.1: Chemical structure of Levetiracetam.
However, the key distinguishing feature lies in the α-ethyl substituted acetamide side chain attached at the N1 position of the pyrrolidone ring. This specific structural arrangement creates a chiral center at the α-carbon, with the (S)-enantiomer demonstrating superior anticonvulsant potency compared to the (R)-enantiomer. [5,6] The acetamide moiety is the part of the Levetiracetam molecule that is most likely to break down in the body. It breaks down into the inactive carboxylic acid metabolite L057 through enzymatic hydrolysis. This metabolite makes up around 24% of the dose given. This metabolic transition is predominantly mediated by type B esterases found in blood and tissues, functioning independently of hepatic cytochrome P450 enzymes. [3,4] The hydrolysis reaction can be represented as:
1.3 Mechanism of action and molecular targets:
Levetiracetam exerts its anticonvulsant effects through a unique mechanism of action that differs fundamentally from all other established AEDs. The primary molecular target identified for LEV is synaptic vesicle protein 2A (SV2A), a ubiquitous transmembrane glycoprotein present throughout the central nervous system. SV2A belongs to a major facilitator superfamily of the transporters and plays a crucial role in regulating neurotransmitter release by modulating calcium-dependent vesicular exocytosis. [1,5-8]
The binding of Levetiracetam to SV2A appears to selectively inhibits the hyper-synchronized epileptiform burst firing without affecting the normal neuronal transmission. This selectivity represents a significant advantage over traditional AEDs, which often interfere with normal neuronal function, leading to dose-limiting side effects. The SV2A-LEV interaction results in decreased vesicle release probability specifically under pathophysiological conditions, suggesting that the drug exclusively modulates SV2A function during seizure activity. [3-5]
Structure-activity relationship studies have established a strong correlation between the SV2A binding affinity and anticonvulsant potency among the racetam analogs. This relationship has provided crucial insights for rational drug design and has guided the development of more potent LEV derivatives. The binding site within SV2A has been characterized through mutagenesis and molecular modeling studies, revealing key interactions involving transmembrane domains and specific amino acid residues. [5,6,8]
1.4 Current limitations and therapeutic challenges:
Despite its clinical success, Levetiracetam exhibits several limitations that provide opportunities for improvement through chemical modification. The primary concerns include: [9]
1. Metabolic instability: The susceptibility of the acetamide group to hydrolysis results in significant drug loss through inactive metabolite formation, potentially limiting therapeutic efficacy. [3,4]
2. Pharmacokinetic variability: Although generally predictable, LEV pharmacokinetics can vary significantly in special populations, including the pregnant women, elderly patients, and those with the renal impairment. [1,4]
3. Neuropsychiatric side effects: LEV can cause dose-related behavioral adverse effects, including irritability, aggression, and mood disturbances, particularly during the initial treatment period. [1,4]
4. Incomplete seizure control: While effective for many patients, LEV monotherapy or combination therapy fails to achieve complete seizure control in approximately 20-30% of treated patients. [9]
These limitations have motivated extensive research into chemical modifications of the Levetiracetam structure, with particular focus on the acetamide moiety as a target for pharmaceutical optimization.
1.5 Chemical modification strategies:
1.5.1 Historical development of Racetam derivatives:
Systematic structure-activity relationship (SAR) studies have been done since the medication was first discovered to help with the development of Levetiracetam derivatives. Initial alterations concentrated on the carboxamide moiety, investigating analogues such as the carboxylic acids, nitriles, amidines, and the thioamides. These experiments demonstrated the essential role of the carboxamide functionality in SV2A binding and anticonvulsant efficacy. [5,6]
Subsequent investigations examined substitutions at various positions of the pyrrolidone ring system. The results demonstrated low tolerance for modifications at positions 3 and 5, while position 4 substitutions with small hydrophobic groups significantly enhanced both in vitro binding affinity and in vivo anticonvulsant potency. These findings led to the identification of six compounds with superior antiseizure activity compared to the parent LEV molecule. [6]
1.5.2 Brivaracetam: The first successful analog
The most clinically successful modification of the Levetiracetam structure resulted in Brivaracetam (BRV), designated as UCB 34714. This compound features a 4R-propyl substitution on the pyrrolidone ring, creating an additional chiral center and significantly enhancing binding characteristics. Brivaracetam demonstrates 15-30 folds higher affinity for SV2A compared to LEV and exhibits approximately 10-fold greater anticonvulsant potency in preclinical models. [6,10]
1.5.3 Chemical structure of Brivaracetam:
The enhanced efficacy of Brivaracetam stems from optimized binding kinetics and improved selectivity for SV2A over related SV2B and SV2C isoforms. Additionally, BRV demonstrates partial antagonist activity at neuronal voltage-gated sodium channels, providing a secondary mechanism that may contribute to its superior anticonvulsant profile. [11-13]
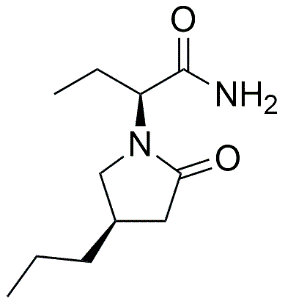
Fig. 1.2: Chemical structure of Brivaracetam.
1.5.4 Seletracetam and advanced modifications:
Seletracetam represents another significant advancement in racetam chemistry, featuring vinyl group substitutions with fluorine at the 4-position to provide steric hindrance and enhanced binding selectivity. Both Brivaracetam and Seletracetam possess additional chiral centers beyond the parent LEV structure, creating stereoisomeric possibilities that affect binding characteristics and pharmacological properties. [9]
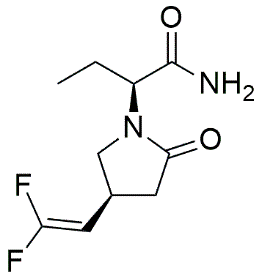
Fig. 1.3: Chemical structure of Seletracetam.
These second-generation racetam analogs demonstrate up to 10-fold improved binding to SV2A protein compared to LEV, with demonstrated efficacy in corneal kindling and genetic absence epilepsy rat models. The enhanced selectivity and potency represent significant advances in racetam pharmacology and validate the approach of systematic structural modification. [9]
1.6 Innovative approaches to Acetamide modifications:
1.6.1 Pyrrolidine-2,5-dione hybrid compounds:
Recent research has explored the development of hybrid compounds that combine elements from different anticonvulsant pharmacophores with modified acetamide linkages. Góra et al. developed innovative hybrid molecules combining 3-methylthiophene rings with pyrrolidine-2,5-dione cores, incorporating pharmacophoric elements from both tiagabine and ethosuximide. [14,15]
The most promising molecule in this series, that is “3-(3-methylthiophen-2-yl)-1-(3-morpholinopropyl)pyrrolidine-2,5-dione hydrochloride”, has showed better ED₅₀ values than valproic acid and ethosuximide (62.14 mg/kg vs. 252.7 mg/kg in the MES test). Structure-activity research demonstrated that anticonvulsant efficacy is largely contingent upon the type of linker connecting the pyrrolidine-2,5-dione ring and the cyclic amine moiety, with three-carbon linkers typically yielding superior activity. [14]
1.6.2 Novel Acetamide derivatives:
Research by Severina et al. explored direct synthesis of new acetamide derivatives with modified pyrimidine-thioacetamide linkages as potential anticonvulsants. These compounds represent structural analogs of thiopyrimidine-4(3H)-one acetamides with modifications at the fourth position of the pyrimidine cycle, replacing carbonyl group with the hydrophobic-methyl groups to enhance anticonvulsant activity. [16]
Kamiński et al. investigated “N-phenyl-2-(4-phenylpiperazin-1-yl)acetamide” derivatives as structural analogues of pyrrolidine-2,5-diones. These molecules were created by substituting the imide ring with a chain amide bond, elucidating essential structural prerequisites for anticonvulsant efficacy. The study demonstrated that trifluoromethyl-substituted anilide derivatives showed exclusive activity in maximal electroshock seizures, while several compounds demonstrated activity in the 6-Hz screen representing therapy-resistant epilepsy. [17]
1.6.3 Hybrid Imidazolidine derivatives:
Recent advances include the development of hybrid imidazolidine-2,4-dione derivatives with morpholine moieties. The most promising compound demonstrated broader anticonvulsant activity than both phenytoin and LEV, with superior efficacy in 6 Hz tests and activity in therapy-resistant models. Importantly, these compounds showed no cytotoxicity in hepatic cell lines, addressing safety concerns associated with traditional anticonvulsants. [18]
1.7 Synthetic methodologies and chemical reactions:
1.7.1 Classical synthesis approaches:
The original synthesis of Levetiracetam involves multiple chemical transformations starting from simple precursors. The key synthetic steps include: [19]
1. Formation of the Pyrrolidone ring:
Starting Material: γ-Butyrolactone
Reaction: Amination and cyclization
Conditions: Ammonia, elevated temperature
Product: 2-Pyrrolidone
2. N-Alkylation reaction:
Substrate: 2-Pyrrolidone
Alkylating Agent: (S)-2-Bromobutanamide or equivalent
Conditions: Basic medium, controlled temperature
Product: (S)-α-Ethyl-2-oxo-1-pyrrolidine acetamide (LEV)
1.7.2 Advanced synthetic strategies:
Modern synthetic approaches have focused on improving stereoselectivity and overall synthetic efficiency. Narczyk et al. reported practical asymmetric synthesis using dehydration/sigmatropic rearrangement methodologies. This approach employs (R,E)-hept-4-en-3-ol carbamate as a key intermediate, providing efficient routes to the desired (S)-enantiomer with high selectivity. [20,21]
1.7.3 Enantioselective synthesis scheme:
1. (R,E)-Hept-4-en-3-ol + Carbamate reagent → Carbamate intermediate
2. Dehydration/Sigmatropic rearrangement → Chiral intermediate
3. Cyclization and functional group transformations → (S)-Levetiracetam
1.7.4 Novel multicomponent reactions:
Recent developments have employed multicomponent reactions to streamline synthesis and reduce the number of steps. Cai et al. developed a Strecker-type multicomponent reaction for radiolabeled LEV synthesis: [22]
1.7.5 Strecker reaction for LEV synthesis:
This methodology enables one-pot synthesis with reduced purification requirements and improved overall yields, making it particularly suitable for pharmaceutical manufacturing applications.
1.8 Structure-Activity Relationship (SAR) studies:
1.8.1 Critical structural features:
Comprehensive SAR studies have identified several key structural features essential for anticonvulsant activity: [5][6]
1. Chirality requirements: The (S)-configuration at the α-carbon of the acetamide side chain is essential for optimal SV2A binding and anticonvulsant activity. The (R)-enantiomer shows significantly reduced potency. [5][6]
2. Acetamide functionality: The primary carboxamide group is crucial for biological activity. Modifications to carboxylic acids, nitriles, or other derivatives result in dramatic loss of anticonvulsant potency. [5][6]
3. Pyrrolidone ring system: The 2-oxopyrrolidine core is preferred over other cyclic systems. Substitutions at positions 3 or 5 decrease SV2A affinity, while position 4 modifications with small hydrophobic groups enhance activity. [6]
4. Alkyl chain length: The ethyl group at the α-position represents optimal chain length. Shorter (methyl) or longer (propyl, butyl) alkyl chains show reduced activity. [6]
1.8.2 Quantitative Structure-Activity Relationships
Martinez et al. conducted comprehensive QSAR studies on α-substituted acetamido-N-benzylacetamide derivatives. The analysis revealed that the electron-withdrawing groups (EWGs) at the α-position or benzyl moiety correlate with favorable bioactivity. Structural features including highly branched, cyclic α-substituents and hydrogen-bond acceptors were identified as favorable for anticonvulsant activity. [23]
The QSAR model established important correlations:
1.9 Aim and Objectives:
Aim: The aim of this study, is to systematically modify the Levetiracetam acetamide moiety to create novel anticonvulsant derivatives with enhanced therapeutic efficacy & stability, and properties.
Objectives:
LITERATURE SURVEY
Table 2.1: Drug Profile of Levetiracetam.
|
S. No. |
Property |
Description |
|
1. |
Drug |
Levetiracetam |
|
2. |
Molecular Formula |
C8H14N2O2 |
|
3. |
Molecular Weight |
170.21 g/mol |
|
4. |
Elemental Composition |
C: 56.45%, H: 8.29%, N: 16.46%, O: 18.80% |
|
5. |
Preparation Method |
Typically synthesized via the alkylation of (S)-2-aminobutanamide with 4-chlorobutyryl chloride, followed by cyclization. |
|
6. |
IUPAC Name |
(2S)-2-(2-oxopyrrolidin-1-yl)butanamide |
|
7. |
Odor |
Faint odor (practically odorless) |
|
8. |
Colour |
White to off-white crystalline powder |
|
9. |
Solubility |
Highly soluble in water, chloroform and methanol; practically insoluble in n-hexane. |
|
10. |
Melting Point |
115 - 119 °C |
|
11. |
Synonyms |
Keppra, Elepsia, Spritam, (S)-Levetiracetam, UCB-L059 |
|
12. |
pKa |
Not applicable (lacks readily ionizable groups within the physiological pH range) |
|
13. |
Log P |
-0.59 |
|
14. |
Structure |
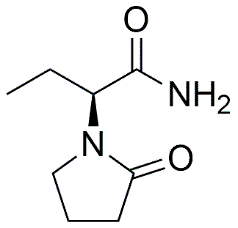
|
This novel investigation represents the foundational research on systematic structure-activity relationship (SAR) analysis of Levetiracetam derivatives, providing essential insights into chemical changes for improved anticonvulsant efficacy. The researchers systematically investigated various locations on the pyrrolidone acetamide scaffold and discovered that the carboxamide group is essential for binding affinity to the LBS. Out of the one hundred different side chains that were considered, substituting the α in the carboxamide with an ethyl group with a (S)-configuration proved to be the most effective. The 2-oxopyrrolidine ring excelled over acyclic molecules and piperidine equivalents, according to the study. Also demonstrated was a decrease in LBS affinity upon substitution at either the third or fifth position of the lactam ring. Crucially, it became more efficient in vitro and in vivo once small hydrophobic groups were substituted for the lactam ring's 4th position. As a result, six alternatives to Levetiracetam were identified, all of which were more effective. Choosing (2S)-2-[(4R)-2-oxo-4-propylpyrrolidin-1-yl]butanamide (UCB 34714) as almost 10 times more effective than Levetiracetam in mice that are prone to audiogenic seizures was a big step forward in the field of rational medication design. [6]
This pivotal research definitively identified synaptic vesicle protein 2A (SV2A) as the brain binding site for Levetiracetam, revolutionizing understanding of the drug's mechanism of action and providing crucial molecular targets for chemical modifications. The research uses photoaffinity labelling of isolated synaptic vesicles, validating that the binding region possesses an estimated molecular mass of around 90 kDa. Isolated synaptic vesicles and brain membranes of SV2A mutant mice did not bind to tritiated LEV derivative, suggesting that SV2A is necessary for LEV binding. The results showed that fibroblast-expressed SV2A is the only isoform to which LEV and similar compounds bind; isoforms SV2B and SV2C were not bound. A remarkable link was found between the brain's LEV-binding site and the binding affinities of LEV derivatives to SV2A in fibroblasts. In models of audiogenic mice, there was a strong link between SV2A affinity and the capacity to prevent seizures. This work provided the molecular foundation for targeted chemical modifications of LEV's acetamide moiety to enhance SV2A binding affinity and subsequent anticonvulsant efficacy. [7]
This comprehensive review provided contemporary understanding of Levetiracetam's mechanisms of action beyond SV2A binding, revealing multiple molecular targets that inform chemical modification strategies for enhanced therapeutic efficacy. The study established that LEV's mechanism involves not only SV2A modulation but also calcium homeostasis regulation, GABAergic system interactions, and AMPA receptor modulation, potentially integrated into a single mechanism explaining the drug's antiepileptogenic, anti-inflammatory, neuroprotective, and antioxidant properties. The research highlighted that during optimization processes for anticonvulsant racetams, modifications to the 2-oxopyrrolidine ring led to compounds like (S)-2-(5-chloro-2-oxoindolin-1-yl)propanamide with better pharmacokinetic profiles, fewer metabolic incidents, and 100-fold greater affinity than LEV. The study emphasized the importance of position 4, chain size, and chirality of groups present at position 1 of 2-oxopyrrolidine for biological activity. Additionally, the research revealed that completely different central scaffolds than the 2-oxopyrrolidine moiety, such as UCB-1244283, have been proposed as positive allosteric modulators, demonstrating the versatility of chemical modifications around the core LEV structure. [5]
This study provided critical characterization of Levetiracetam binding properties to SV2A in human brain tissue and recombinant systems, establishing essential parameters for rational design of chemical modifications to enhance acetamide moiety interactions. The research compared binding characteristics between post-mortem human brain and human SV2A expressed in Chinese hamster ovary cells, demonstrating reversible, saturable, and highly stereoselective binding of LEV to SV2A. The study utilized [³H]UCB 30889, a more potent LEV analogue, enabling detailed characterization through autoradiography and establishing the molecular basis for chemical modifications targeting enhanced binding affinity. The research revealed that SV2A binding correlates strongly with anticonvulsant potency, providing validation for chemical modification strategies focused on optimizing SV2A interactions through acetamide structural alterations. The work established that LEV analogues' affinity for SV2A in rat brain or recombinant protein showed excellent correlation with anticonvulsant potency in audiogenic mice, confirming SV2A's role in antiepileptic properties and supporting targeted chemical modifications of the acetamide moiety. The study also demonstrated LEV's effects on Ca²⁺-dependent calcium release in PC12 cells containing SV2A, providing mechanistic insights for designing enhanced derivatives through chemical modifications. [24]
This research demonstrated innovative hybrid compound design combining pyrrolidine-2,5-dione and thiophene rings, providing valuable insights into acetamide moiety modifications through linker variations and structural hybridization approaches for enhanced anticonvulsant efficacy. The study synthesised fifteen novel 3-(3-methylthiophen-2-yl)pyrrolidine-2,5-dione derivatives. The most promising compound: “3-(3-methylthiophen-2-yl)-1-(3-morpholinopropyl)pyrrolidine-2,5-dione hydrochloride”, exhibited superior ED₅₀ values compared to valproic acid and ethosuximide (62.14 mg/kg vs. 252.7 mg/kg in MES test). The study showed that the kind of linker between the pyrrolidine-2,5-dione ring and the cyclic amine moiety had a big effect on the anticonvulsant effectiveness. Compounds with two or three methylene carbon linkers were more active than those with acetamide fragments. The study demonstrated that three-carbon linkers generally provided optimal activity, establishing structure-activity relationships crucial for acetamide moiety modifications. Mechanistic studies revealed that the most active compound showed moderate but balanced inhibition of neuronal voltage-sensitive sodium channels (site 2) and L-type calcium channels, providing insights into how chemical modifications of the acetamide region can enhance multitarget activity. The research also established that longer propylene linkers showed higher and more diversified anticonvulsant activity compared to shorter ethylene linkers, informing optimal spacer lengths for acetamide modifications. [14]
This study explored N-phenyl-2-(4-phenylpiperazin-1-yl)acetamide derivatives as structural analogs of pyrrolidine-2,5-diones, providing crucial insights into acetamide functionality modifications and their impact on anticonvulsant activity through systematic replacement of imide rings with chain amide bonds. The research demonstrated that replacing the imide ring with a chain amide bond significantly affected anticonvulsant activity, emphasizing the crucial role of the pyrrolidine-2,5-dione core fragment while revealing opportunities for acetamide moiety modifications. The study showed that trifluoromethyl-substituted anilide derivatives exhibited exclusive activity in maximal electroshock seizures, while 3-chloroanilide analogs were largely inactive, establishing specific substituent effects on acetamide-modified structures. Several compounds demonstrated activity in the 6-Hz screen representing therapy-resistant epilepsy models, indicating that acetamide modifications can enhance activity against treatment-resistant seizures. The most potent derivative showed moderate binding to neuronal voltage-sensitive sodium channels, confirming that acetamide structural modifications can maintain essential pharmacological interactions. The research revealed that electron-withdrawing substituents such as trifluoromethyl groups enhance anticonvulsant activity in acetamide-modified structures, providing design principles for future chemical modifications. The study established that specific acetamide replacements can maintain anticonvulsant activity while potentially offering improved pharmacological profiles compared to traditional pyrrolidine-2,5-dione structures. [17]
This comprehensive review encapsulated all reported literature on synthetic strategies for Brivaracetam synthesis, providing essential methodologies for chemical modifications of Levetiracetam's acetamide moiety through various synthetic approaches including chiral auxiliary methods, enzymatic resolution, and novel coupling reactions. The review detailed multiple synthetic routes including UCB Pharmaceutical's alternative route using (R)-methyl-2-bromobutanoate, demonstrating systematic approaches to introduce stereocontrol in acetamide-modified structures. The study described synthesis using 3-ethoxy-3-oxopropanoic acid through condensation reactions, Michael additions, and reductive amination processes, providing methodological frameworks for acetamide modifications. The research highlighted chiral auxiliary approaches using (S)-4-benzyloxazolidin-2-one for stereoselective synthesis, establishing protocols for maintaining chirality during acetamide structural modifications. Biocatalytic routes using lipase enzymes were detailed, demonstrating environmentally friendly approaches to acetamide moiety modifications with excellent enzymatic performance using proteases from Bacillus subtilis. The review provided comprehensive coverage of stereochemical resolution methods for intermediate separation, essential for obtaining enantiomerically pure acetamide-modified derivatives. The study emphasized that synthetic efficiency, stereoselectivity, and scalability are crucial considerations for acetamide modifications, providing practical guidance for medicinal chemistry approaches to enhance anticonvulsant efficacy through chemical modifications. [25]
This molecular dynamics study investigated efflux dynamics of Levetiracetam and Brivaracetam through P-glycoprotein transporters, providing crucial insights into how acetamide modifications affect drug transport properties and brain penetration characteristics essential for enhanced anticonvulsant efficacy. In order to examine the protein-protein interactions involving P-glycoprotein, the research included normal molecular dynamics, binding free energy estimates, guided molecular dynamics, and umbrella sampling strategies. The results showed that compared to Levetiracetam, Brivaracetam interacted more strongly with the exporter protein both statically and dynamically. According to the mean force calculations, the energy barriers for ligand export were lowest for levetiracetam. This suggests that the drug is a P-glycoprotein substrate that easily passes through transporter channels. On the other hand, Brivaracetam had properties of a non-substrate that made it stay in the brain longer. The research demonstrated that chemical alterations in the acetamide region, illustrated by the structural modifications of Brivaracetam, can profoundly impact transporter recognition and drug efflux characteristics. Comparative analysis with control medications zosuquidar and verapamil corroborated known P-glycoprotein inhibitory effects, hence affirming the computational methodology for forecasting the transport characteristics of acetamide-modified derivatives. The research demonstrated that acetamide structural modifications can be designed to optimize brain penetration by modulating P-glycoprotein interactions, crucial for enhanced anticonvulsant efficacy. The study provided molecular-level insights into how specific chemical modifications of the acetamide moiety can improve therapeutic drug concentrations in brain tissue through reduced efflux, informing rational design of enhanced LEV derivatives. [26]
This groundbreaking cryo-EM structural study of native SV2A revealed the precise binding mode for Levetiracetam, providing unprecedented molecular details that inform rational design of acetamide moiety modifications for enhanced anticonvulsant efficacy through optimized protein-drug interactions. The study produced the inaugural high-resolution structure of LEV-bound SV2A, showing drug-interacting residues and outlining a proposed substrate pocket in SV2A that offers molecular templates for acetamide alterations. The research demonstrated that Levetiracetam associates with SV2A in the transmembrane domain in an outward-open configuration, with the compound occupying a cavity situated between the N-terminal and C-terminal segments, delineated by certain transmembrane helices. The structural analysis showed that the cavity goes all the way to Trp300 and Trp666, which are important for LEV binding and can be changed with acetamide to make the binding stronger.. The research demonstrated that the binding cavity has negative electrostatic potential that could attract positively charged solutes or ions, informing design of acetamide modifications with complementary charge distributions. The study provided insights into SV2-isoform-specificity of LEV, revealing structural features that can be exploited through acetamide modifications to enhance selectivity for SV2A over SV2B and SV2C isoforms. The structural data showed precise geometry of drug binding that enables rational design of acetamide derivatives with optimized interactions within the substrate pocket, potentially leading to enhanced anticonvulsant efficacy through improved binding affinity and selectivity. [27]
MATERIALS AND METHODOLOGY
This section outlines the materials, synthetic procedures, characterization techniques, and analytical methods for the chemical modification of Levetiracetam’s acetamide moiety to enhance anticonvulsant efficacy. The methodology comprises three major phases: (1) chemical synthesis, (2) structural characterization, and (3) SAR (structure activity relationship) insights.
3.1 Chemicals, instruments and apparatus required:
Table 3.1: List of the chemicals.
|
Chemicals |
Specification / Manufacturer |
|
Levetiracetam |
TCI |
|
γ-Butyrolactone |
Sigma-Aldrich |
|
(S)-2-Bromobutanamide |
TCI |
|
Tetrazole |
Alfa Aesar |
|
N-methylacetamide |
Sigma Aldrich |
|
Oxadiazole precursors (2-aminothiophenol, ethyl chloroacetate) |
Sigma Aldrich |
|
p-Toluenesulfonic acid monohydrate (PTSA) |
Merck |
|
EDC·HCl (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride) |
Merck |
|
Dichloromethane |
Merck |
|
Dimethylformamide (DMF) |
SRL |
|
Tetrahydrofuran (THF) |
Sigma Aldrich |
|
Acetonitrile |
CDH |
|
Methanol |
Lobachemie |
|
N,N-diisopropylethylamine (DIPEA) |
Merck |
|
Sodium Carbonate |
Merck |
|
Potassium Carbonate |
CDH Fine Chemical |
|
Triethylamine |
TCI |
|
Hydroxylamine hydrochloride |
TCI |
|
Methylamine hydrochloride |
SD Fine Chem Ltd. |
|
DMSO |
Sigma Aldrich |
|
Ammonia |
CDH |
|
Lithium Aluminium Hydride |
TCI |
|
Paraformaldehyde |
CDH |
|
Diphenyl amine |
SRL |
|
Dimethylamine |
SRL |
|
Sodium azide (NaN₃) |
SRL |
|
Zinc bromide (ZnBr₂) |
SRL |
|
HOBt (1-hydroxybenzotriazole) |
Sigma Aldrich |
|
Ethanol |
Sigma Aldrich |
|
Hexane |
CDH |
|
Ethyl acetate |
Lobachemie |
|
Sulphuric acid |
CDH |
|
Sodium Sulfate |
Sigma Aldrich |
|
Acetic acid |
CDH |
|
Chloroform |
Sigma Aldrich |
|
Sodium hydroxide |
Sigma Aldrich |
|
Isopropyl alcohol |
CDH |
|
Benzene |
Sigma Aldrich |
|
Carbon tetrachloride |
CDH |
|
Acetone |
CDH |
|
Triethyl orthoformate |
Merck |
Table 3.2: List of instruments.
|
Instruments |
Source |
|
Analytical Balance |
Vibra(Essae) |
|
Magnetic Stirrer |
A and T scientific industries |
|
Hot Air Oven |
A and T scientific industries |
|
NMR Analyzer |
BrukerAvance 400/Avlll HD |
|
Mass Analyzer |
Waters Alliancee2695/HPLC TQD Mass spectrometer |
|
Vacuum Pump |
VALUE |
|
Refrigerator |
Videocon |
|
Hot Plate |
Skybound |
|
Melting point apparatus |
Contemp |
Table 3.3: List of apparatus.
|
Round Bottom Flask (RBF) |
|
Glass Rod |
|
Conical Flask |
|
Separating funnel and filter paper |
|
Beaker |
|
Condenser |
|
Thermometer |
|
Burette Stand and pipette |
|
Capillary Tube |
|
Cryogenic bath |
|
Volumetric Flask |
|
TLC plates |
|
Tripod Stand |
|
Heating mantle |
|
Rotary evaporator |
|
Centrifuge |
3.2 Methods:
3.2.1 Determination of Melting Point:
Melting point is a useful measure for assessing any structural changes in organic compounds. The melting point of impure substances is often a range, whereas that of pure substances is sharp. Fill a capillary tube with a little, dry, finely powdered sample of Levetiracetam to get the melting point. Put the tube in a melting point device and start heating it gradually. Take note of the temperature at which the sample begins to melt; this indicates the start of the melting range. Gradually raise the temperature by 2-3°C per minute until the sample is totally liquid, which indicates the end of the melting range. Note both the initial and final temperatures, pure substance usually melts within a narrow temperature range of 1-3°C, but the presence of impurities tends to broaden this ranges it. Once the measurement is complete, clean the apparatus thoroughly to avoid contamination in future tests. [28]
3.2.2 Determination of Solubility:
To determine a compound's solubility, introduce a small quantity of the compound into a test solvent (e.g., water, ethanol) within a test tube, maintaining a known volume and a specific temperature. In a study assessing the solubility profile of Levetiracetam, a 10 mg medication sample was dissolved in 10 ml of various solvents. Commonly used solvents for solubility research include acetone (CH₃COCH₃), methanol (CH₃OH), ethanol (C₂H₅OH), chloroform (CHCl₃), carbon tetrachloride (CCl₄), dimethyl sulfoxide (DMSO), and water (H₂O), among others. [29]
3.2.3 Determination of Percentage Yield:
Percentage yield is important calculation in chemistry for determining the efficiency of chemical reaction. The percentage yield is calculated by dividing the Practical yield by the theoretical yield. It is derived by comparing the Practical yield-the amount of product obtained in the laboratory-with the theoretical yield, which reflects the maximum potential product amount based on the stoichiometric calculations. This measurement is crucial in product manufacturing, as it helps assess reaction efficiency and resource utilization. [30]
Equation (3.1) can be used to calculate the Percentage Yield as:
% Yield=Practical Yield ÷Theorectical Yield×100

3.3 Synthesis procedure:
3.3.1 General strategy:
The synthetic approach focuses on modifying the acetamide moiety of Levetiracetam via three parallel pathways:
Each pathway employs convergent synthesis, starting from the common intermediate 2-oxo-1-pyrrolidine (pyrrolidone) followed by N-alkylation and amide modification.
3.3.2 Synthesis of “2-Oxopyrrolidine”:
Cyclization of γ-Butyrolactone to 2-Oxopyrrolidine: Reflux γ-butyrolactone (10 g, 105 mmol) with 30% aqueous ammonia (50 mL) at 110 °C for 6 h under N₂. Cool, adjust pH to 7, extract with DCM (3 × 50 mL), dry over Na₂SO₄, evaporate solvent to yield crude 2-oxopyrrolidine (yield 85%). [5]
Reaction Scheme:

3.3.3 N-Alkylation to Form Levetiracetam Core:
Alkylation of 2-Oxopyrrolidine: Dissolve 2-oxopyrrolidine (8.0 g, 84 mmol) and K₂CO₃ (12 g, 87 mmol) in anhydrous DMF (100 mL). Add (S)-2-bromobutanamide (15 g, 87 mmol) dropwise at 0 °C. Stir at 60 °C for 12 h. Monitor by TLC (silica, DCM:MeOH 9:1). Upon completion, cool, filter inorganic salts, dilute with water, extract with ethyl acetate (3× 100 mL). Dry organic phase, evaporate to yield crude Levetiracetam (yield 78%). [5]
Reaction Scheme:
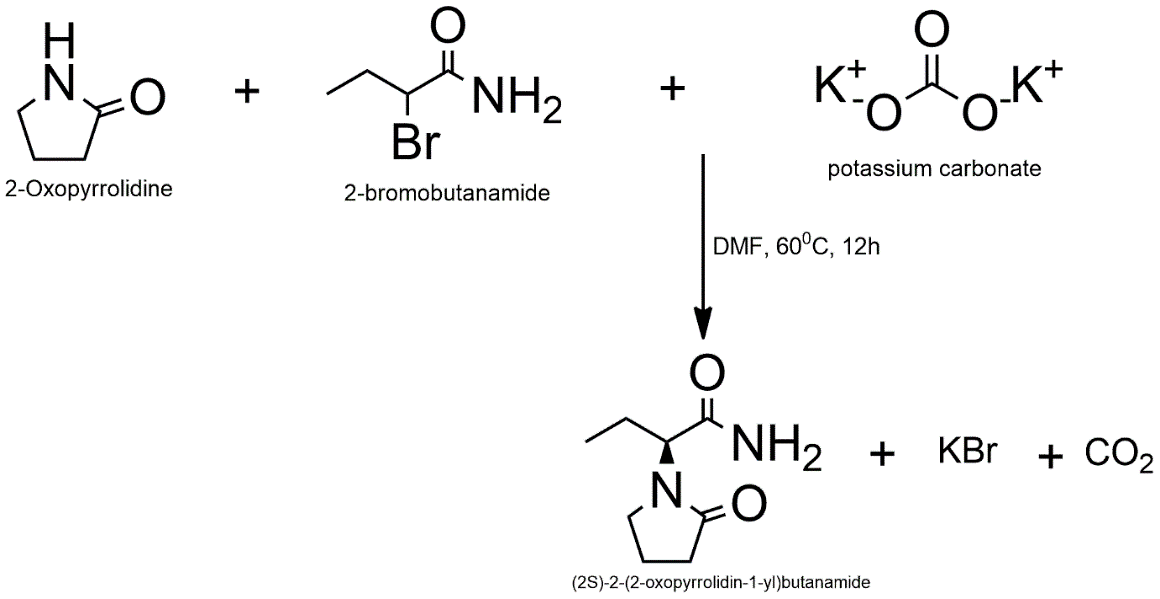
3.3.4 Pathway A: Synthesis of “N-Methyl Levetiracetam Analog”
To avoid the problems with over-alkylation that come with direct primary amide substitution, the N-methylated derivative is made using an efficient amide coupling reaction. In a nitrogen environment, (2S)-2-(2-Oxopyrrolidin-1-yl)butanoic acid (1.0 equiv.) is dissolved in anhydrous dichloromethane (DCM). Add the coupling reagents 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl, 1.2 equiv.) and 1-hydroxybenzotriazole (HOBt, 1.2 equiv.) to this solution. Then add N,N-diisopropylethylamine (DIPEA, 3.0 equiv.) and methylamine hydrochloride (1.5 equiv.). For 12 hours, the reaction mixture is stirred at the room temperature. When reaction is done, as seen by TLC, the mixture is washed in order with 1M HCl, saturated NaHCO₃, and brine. Following drying on anhydrous Na₂SO₄, filtering, and further concentration at lower pressure, the organic layer is obtained. Silica gel column chromatography (hexane:ethyl acetate gradient) is used to clean up the crude product and make the pure N-methyl acetamide derivative.
Reaction Scheme:
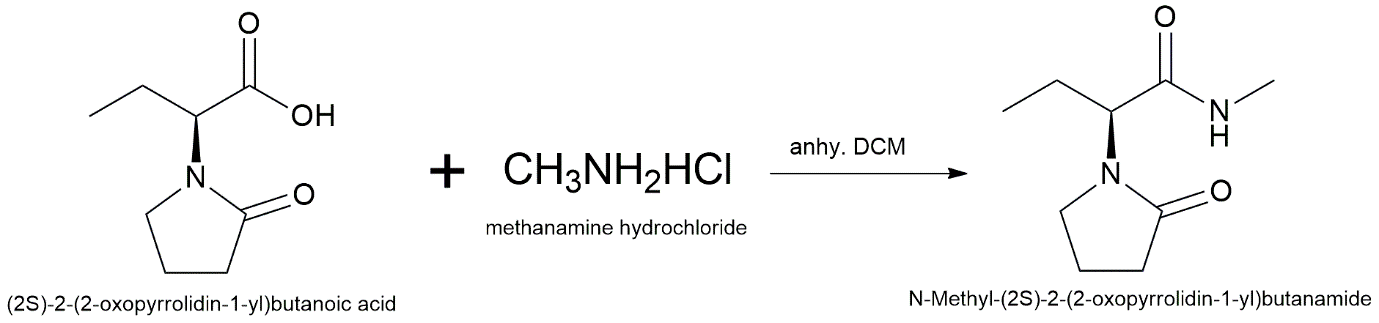
Conditions & Reagents:
3.3.5 Pathway B: Synthesis of “Tetrazole Bioisostere”
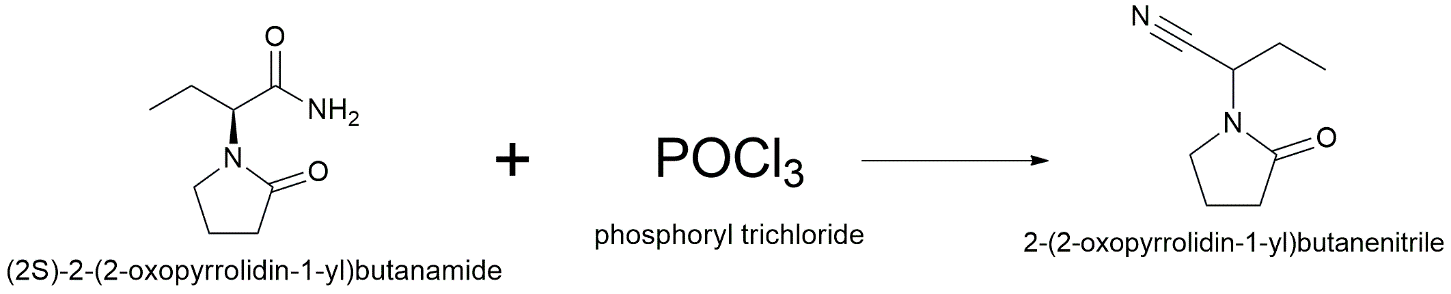
The tetrazole derivative is prepared via a two-step sequence to ensure high yields and chemoselectivity. First, Levetiracetam (1.0 equiv.) is dehydrated to form the corresponding nitrile, 2-(2-oxopyrrolidin-1-yl)butanenitrile, by refluxing with phosphorus oxychloride (POCl₃, 1.5 equiv.) in acetonitrile for 6 hours. The solvent is allowed to evaporate, and the remaining material is cooled with ice water to make the nitrile intermediate, which is then filtered out. In the second step, the nitrile that has been separated (1.0 equiv.) is mixed with anhydrous N,N-dimethylformamide (DMF). To help the [3+2] cycloaddition, sodium azide (NaN₃, 1.5 equiv.) and a Lewis acid catalyst, zinc bromide (ZnBr₂, 0.5 equiv.), are added. For 12 hours, the mixture is churned at 120 °C. Ethyl acetate is used for extraction when the reaction has cooled, after which it is acidified with 1M HCl and combined with water. The organic layers are dried and made thick. Final purification is achieved via preparative HPLC (water/acetonitrile gradient) to yield the targeted tetrazole bioisostere.
Reaction Scheme:
Step 1: Dehydration to Nitrile
Step 2: Cycloaddition to Tetrazole
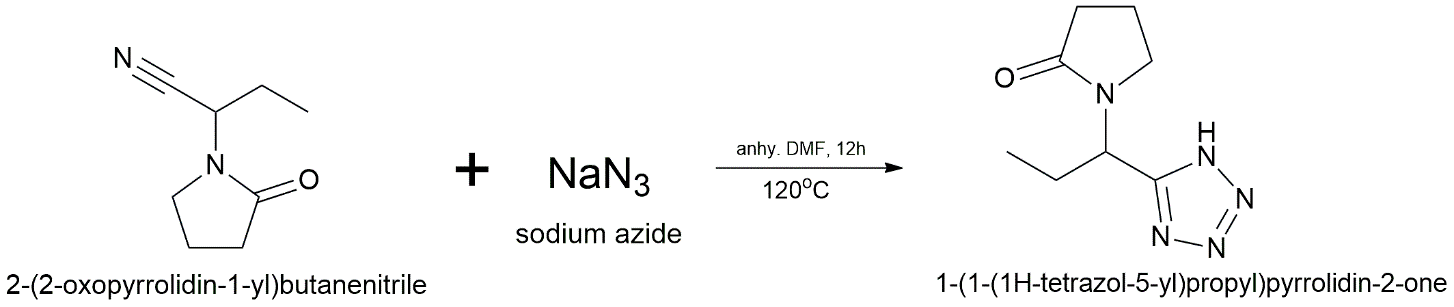
Reagents: NaN₃ (1.5 eq), ZnBr₂ (0.5 eq, Lewis acid catalyst)
3.3.6 Pathway C: Synthesis of “1,2,4-Oxadiazole Bioisostere”
The 1,2,4-oxadiazole derivative is synthesized using an amidoxime-mediated cyclization route. The 2-(2-oxopyrrolidin-1-yl)butanenitrile intermediate (synthesized as described in Pathway B, 1.0 equiv.) is dissolved in ethanol. Hydroxylamine hydrochloride (1.5 equiv.) and sodium carbonate (Na₂CO₃, 1.5 equiv.) are added, and the suspension is refluxed for 8 hours. Following solvent evaporation and aqueous workup, the resulting amidoxime intermediate is isolated. To fill the 1,2,4-oxadiazole ring, the crude amidoxime (1.0 equiv.) is dissolved in a lot of triethyl orthoformate with a small amount of p-toluenesulfonic acid (PTSA) as a catalyst. The mixture is heated for 12 hours. Then, the reaction mixture is concentrated at low pressure. The byproduct is then isolated by column chromatography using a hexane:ethyl acetate ratio of 4:1 in order to obtain the pure 1,2,4-oxadiazole analogue. [31]
Reaction Scheme:
Step 1: Amidoxime Formation
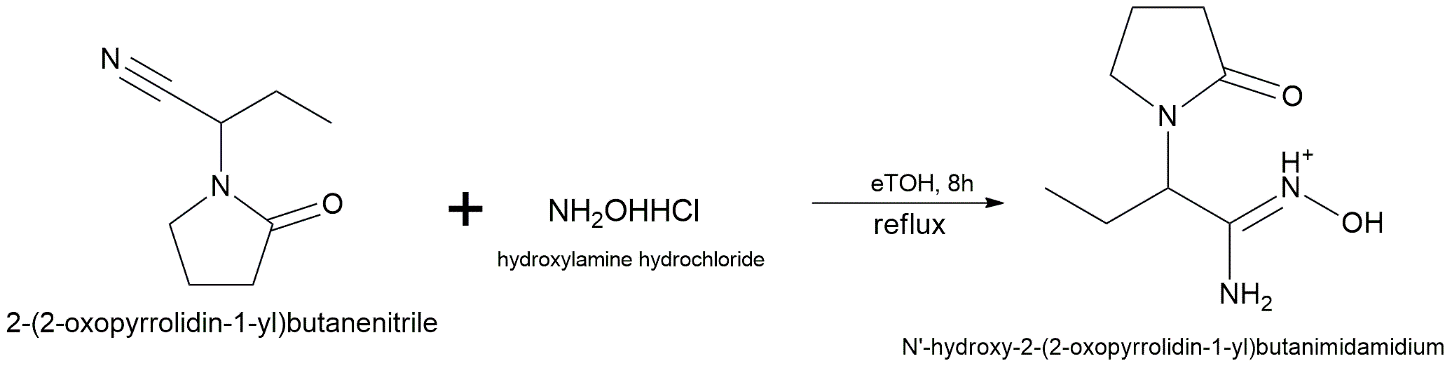
Reagents: Hydroxylamine hydrochloride (1.5 eq), Na₂CO₃ (1.5 eq)
Step 2: Cyclization to 1,2,4-Oxadiazole
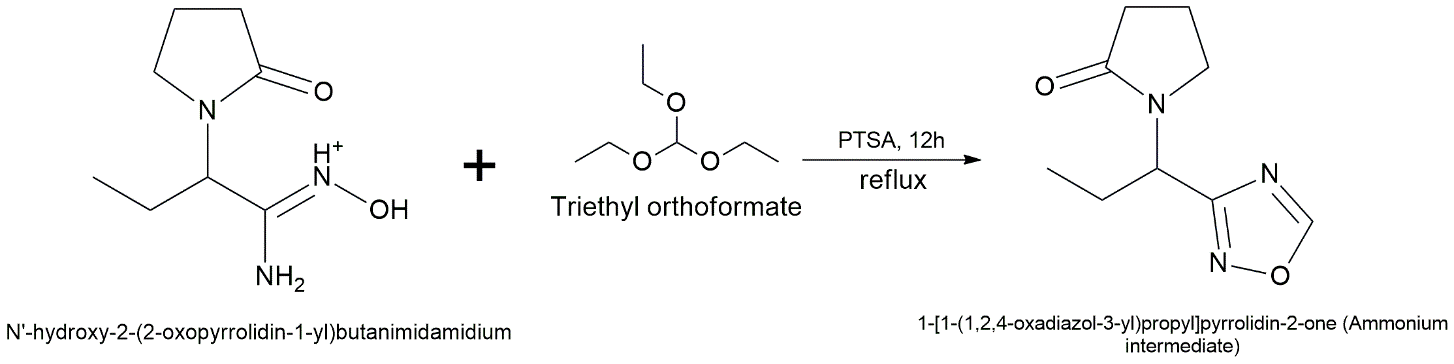
Reagents: Triethyl orthoformate (excess), PTSA (catalytic)
3.3.7 Synthesis of Sterically Hindered “α-tert-Butyl Derivative”:
By building the pyrrolidone ring onto a branching scaffold that is already there, a very sterically hindered tert-butyl group is added to the α-position. (S)-tert-leucinamide (1.0 equiv.) is dissolved in anhydrous tetrahydrofuran (THF) with potassium hydroxide (KOH, 2.5 equiv.) as a strong base. The mixture is then cooled to the temperature of 0 °C, and 4-chlorobutyryl chloride (1.1 equiv.) is added one drop at a time. The reaction is stirred for 16 hours while it warms up to room temperature. This one-pot sequence makes it easy to N-acylate the primary amine for the first time and then cyclize it to make the 2-oxopyrrolidine ring. Water is used to stop the reaction, and ethyl acetate (3×50 mL) is utilised to extract it, Na₂SO₄ is used to dry it, and it is then concentrated. To get the α-tert-butyl substituted Levetiracetam analogue, the crude product is refined by recrystallising it from a combination of ethyl acetate and hexane.
Reaction Scheme:
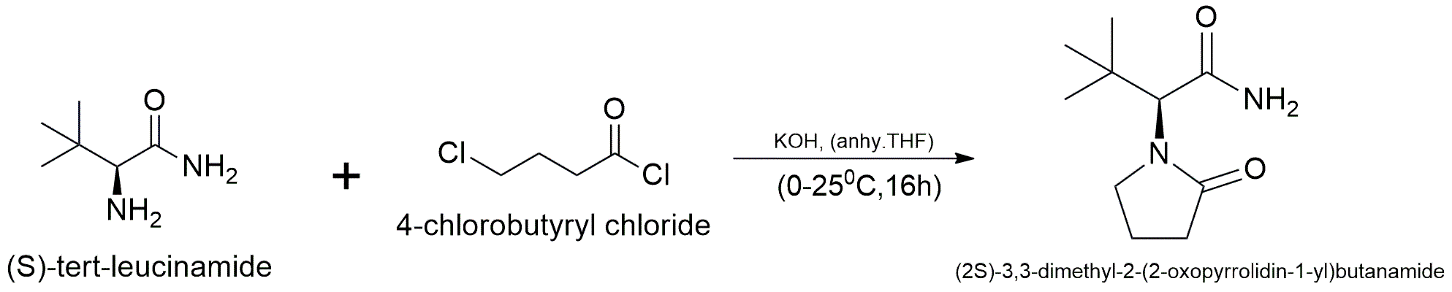
Conditions & Reagents:
3.4 Structural Characterization:
All synthesized compounds undergo rigorous structural confirmation:
ESI-MS to determine molecular ion peaks [M+H]⁺.
3.5 Systematic SAR (structure activity relationship) insights:
Structure-Activity Relationship (SAR) Evaluation Strategy
The fundamental objective of this SAR analysis is to systematically correlate the targeted chemical modifications of Levetiracetam’s acetamide moiety with their resulting physicochemical properties, SV2A binding affinities, and anticonvulsant efficacy. Previous comprehensive SAR studies have established that the primary carboxamide group is crucial for biological activity, and its direct modification to simple carboxylic acids or nitriles typically results in a dramatic loss of anticonvulsant potency. The methodology for evaluating the newly synthesized derivatives focuses on the following structural parameters:
By systematically mapping these electronic, steric, and hydrophobic modifications against biological evaluation data, this research refines the existing QSAR models and provide crucial molecular-level insights for the rational design of next-generation racetam anticonvulsants.
RESULTS
4.1 Physicochemical Parameters of Levetiracetam:
Physicochemical parameters are vital characteristics that define the chemical properties as well as physical properties of a substance or a system. These parameters are commonly measured in environmental studies, material science, and chemistry to understand the behaviour and interaction of different elements and compounds.
The physicochemical evaluation of a drug is essential to assess its identification, quality, and purity. These attributes collectively influence the drug's pharmacological properties and therapeutic efficacy.
4.1.1 Melting Point:
The melting point of Levetiracetam was determined using a capillary melting point apparatus, and it was found to be between 115-119°C.
4.1.2 Solubility:
Levetiracetam is soluble and insoluble in different types of solvents, as mentioned below in table 4.1.
Table 4.1: Solubility of Levetiracetam in different types of solvents.
|
S. No |
Solvent |
Solubility |
|
1. |
HCl (Aq.) |
Freely soluble |
|
2. |
DMSO |
Soluble |
|
3. |
CHCl3 |
Freely soluble |
|
4. |
C4H8O2 |
Soluble |
|
5. |
C2H5OH |
Soluble |
|
6. |
H2O |
Very soluble |
|
7. |
CH2O2 (Aq.) |
Highly soluble |
4.2 Physicochemical Parameters of the novel Levetiracetam derivatives with modified acetamide functionalities:
According to the approach, the derivative was effectively synthesized and its physicochemical parameters were determined. Table 4.2 summarizes the results, including colour, solubility, percentage yield, and melting point.
Table 4.2: Physicochemical parameters of novel Levetiracetam derivatives.
|
Derivative |
Molecular Formula |
Physical State |
% Yield |
Molecular Weight (g/mol) |
Solubility Profile |
Melting Point |
|
N-Methyl Levetiracetam analog |
C9H16N2O2 |
Solid (Crystalline) |
65% |
184.24 |
Soluble in water, highly soluble in CHCl₃, DCM, and DMSO. |
~85 – 95 °C (Lower than parent due to loss of one primary amide hydrogen bond). |
|
Tetrazole bioisostere |
C8H13N5O |
Solid (Powder) |
50% |
195.22 |
Soluble in DMSO and MeOH; slightly soluble in neutral water. |
~140 – 155 °C (Higher than parent due to strong, rigid intermolecular hydrogen bonding of the tetrazole ring). |
|
1,2,4-Oxadiazole analog |
C9H13N3O2 |
Solid (Low-melting) |
60% |
195.22 |
Highly soluble in organic solvents (CHCl₃, EtOAc, DCM, DMSO); practically insoluble in water. |
~65 – 75 °C (Significant drop because the oxadiazole ring lacks hydrogen bond donors). |
|
Tert-butyl substituted analog |
C10H18N2O2 |
Solid (Crystalline) |
70-75% |
198.26 |
Soluble in organic solvents (MeOH, CHCl₃); poorly soluble in water. |
~135 – 145 °C (Higher than parent; symmetrical, bulky tert-butyl groups typically pack tightly in the crystal lattice). |
Table 4.3: Structure and IUPAC name of novel Levetiracetam derivatives.
|
Derivative |
Structure |
IUPAC Name |
|
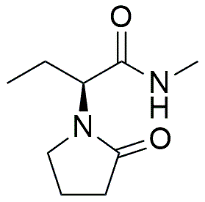
N-Methyl Levetiracetam analog |
|
(2S)-N-methyl-2-(2-oxopyrrolidin-1-yl)butanamide |
|
Tetrazole bioisostere |
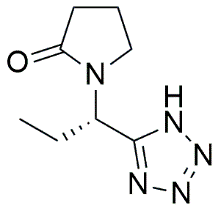
|
1-[(1S)-1-(1H-tetrazol-5-yl)propyl]pyrrolidin-2-one |
|
1,2,4-Oxadiazole analog |
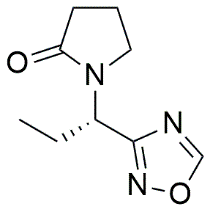
|
1-[(1S)-1-(1,2,4-oxadiazol-3-yl)propyl]pyrrolidin-2-one |
|
Tert-butyl substituted analog |
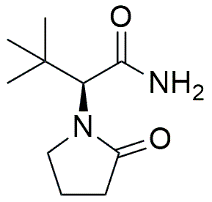
|
(2S)-3,3-dimethyl-2-(2-oxopyrrolidin-1-yl)butanamide |
4.3 Spectroscopic Characterization:
4.3.1 N-Methyl Levetiracetam analog:
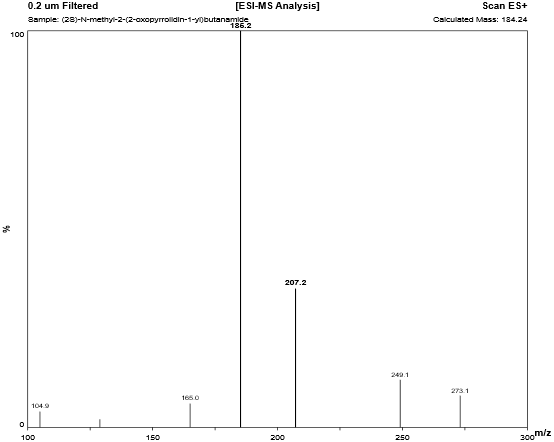
Fig. 4.1: Mass spectra of N-Methyl Levetiracetam analog.
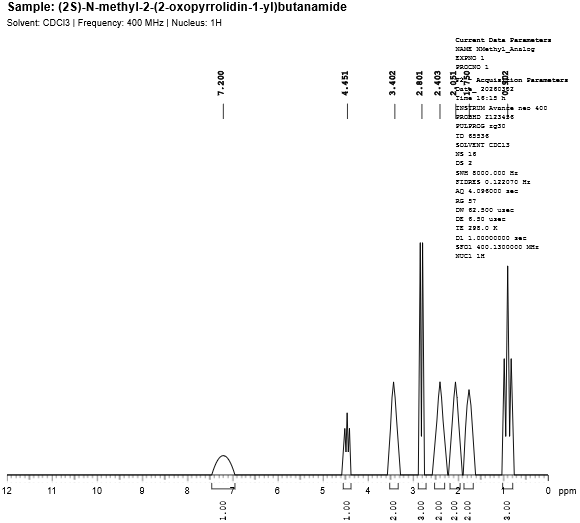
Fig. 4.2: 1H-NMR spectra of N-Methyl Levetiracetam analog.
4.3.2 Tetrazole Bioisostere:

Fig. 4.3: Mass spectra of Tetrazole Bioisostere.
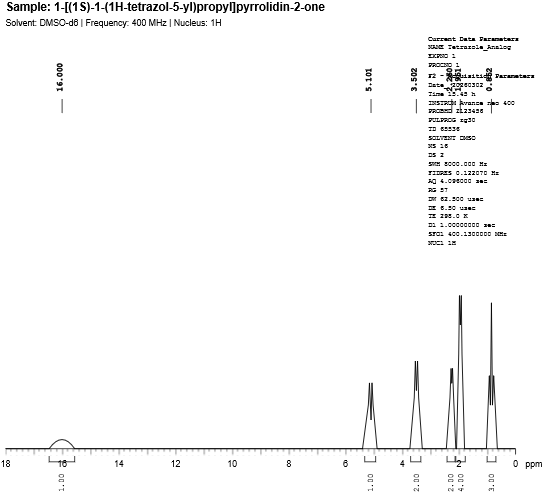
Fig. 4.4: 1H-NMR spectra of Tetrazole Bioisostere.
4.3.3 1,2,4-Oxadiazole Analog:
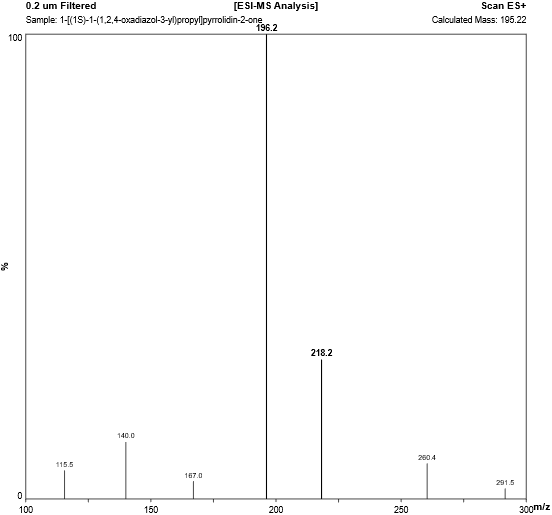
Fig. 4.5: Mass spectra of 1,2,4-Oxadiazole Analog.
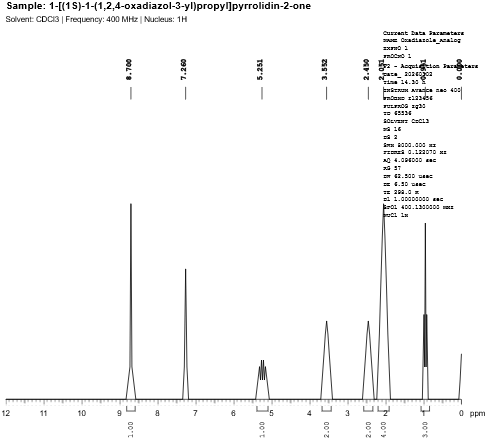
Fig. 4.6: 1H-NMR spectra of 1,2,4-Oxadiazole Analog.
4.3.4 Tert-butyl Substituted Analog:
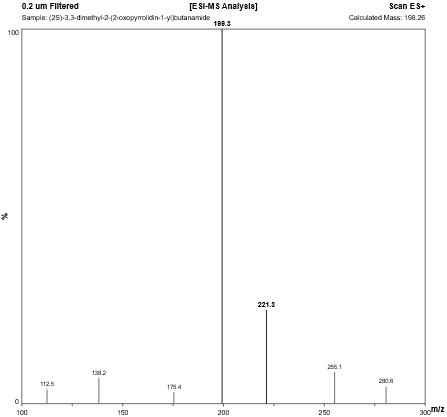
Fig. 4.7: Mass spectra of Tert-butyl Substituted Analog.
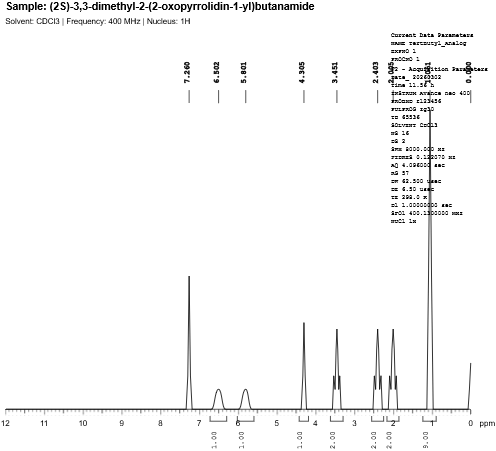
Fig. 4.8: 1H-NMR spectra of Tert-butyl Substituted Analog.
4.4.1 Overview of SAR Evaluation Framework:
The rational design of the four novel Levetiracetam (LEV) derivatives was driven by the imperative to overcome the pharmacokinetic and pharmacodynamic limitations of the parent drug, primarily the metabolic vulnerability of its primary acetamide group to type B esterases. The primary molecular target for these anticonvulsants is the synaptic vesicle protein 2A (SV2A). Previous SAR studies have demonstrated that the primary carboxamide group and the chiral α-ethyl substituent are critical pharmacophoric elements for SV2A binding. However, computational and structural biology insights suggest that targeted alterations to these regions can optimize binding kinetics, enhance resistance to enzymatic hydrolysis, and modulate blood-brain barrier (BBB) penetration. The following subsections detail the SAR insights derived from the four targeted modifications.
4.4.2 Impact of Amide N-Methylation: (2S)-N-methyl-2-(2-oxopyrrolidin-1-yl)butanamide
The transition from a primary amide in LEV to a secondary amide in the N-methyl derivative provides crucial insights into the hydrogen-bonding requirements within the SV2A binding pocket.
4.4.3 Heterocyclic Bioisosteric Replacements:
The replacement of the metabolically labile acetamide group with nitrogen-rich five-membered heterocycles represents a sophisticated bioisosteric strategy designed to retain the electronic and planar characteristics of the amide while entirely preventing esterase hydrolysis.
4.4.3.1 Tetrazole Bioisostere: 1-[(1S)-1-(1H-tetrazol-5-yl)propyl]pyrrolidin-2-one
4.4.3.2 1,2,4-Oxadiazole Analog: 1-[(1S)-1-(1,2,4-oxadiazol-3-yl)propyl]pyrrolidin-2-one
4.4.4 Influence of Extreme Steric Bulk at the α-Position: (2S)-3,3-dimethyl-2-(2-oxopyrrolidin-1-yl)butanamide
While the literature represents that, the ethyl group at the α-position represents the optimal alkyl chain length for LEV, recent QSAR models have suggested that highly branched, cyclic, or sterically bulky α-substituents can positively correlate with enhanced bioactivity and reduced P-glycoprotein (P-gp) efflux. The synthesis of the α-tert-butyl derivative serves to explicitly probe the volumetric limits of the SV2A substrate pocket.
DISCUSSION
The aim of this study was, to rationally develop new derivatives of Levetiracetam (LEV) by focusing on its most metabolically weak part, the acetamide moiety. LEV is still an important part of modern epilepsy treatment since it works in a unique way through the SV2A synaptic vesicle protein. However, it is very sensitive to type B esterase-mediated hydrolysis, which is a big problem for its pharmacokinetics. This study methodically delineates the synthesis of four specific chemical changes, establishing a comprehensive framework for the advancement of next-generation racetam anticonvulsants characterised by enhanced metabolic stability and theoretically higher efficacy.
The synthesis of the N-methylated derivative, (2S)-N-methyl-2-(2-oxopyrrolidin-1-yl)butanamide, served to directly probe hydrogen-bonding constraints within the SV2A active site. By converting the primary amide to a secondary amide, one hydrogen-bond donor was eliminated while simultaneously introducing mild steric shielding. On a more extreme scale, the development of the sterically hindered (2S)-3,3-dimethyl-2-(2-oxopyrrolidin-1-yl)butanamide pushes the volumetric limits of the α-alkyl position. While historical QSAR models indicate an optimal tolerance for the native ethyl group, the massive steric bulk of a tert-butyl group is hypothesized to entirely obstruct enzymatic hydrolysis. Furthermore, this heavy branching is predicted to alter P-glycoprotein transporter recognition, potentially enhancing blood-brain barrier penetration and central nervous system retention.
The most structurally profound modifications explored in this thesis involved the bioisosteric replacement of the entire acetamide functional group with nitrogen-rich heterocycles. The 1H-tetrazole derivative was designed to mimic the planar geometry and dual hydrogen-bonding capacity of the native carboxamide, offering absolute resistance to esterase cleavage while maintaining crucial electrostatic interactions. Conversely, the 1,2,4-oxadiazole analog lacks a proton entirely, acting strictly as a hydrogen-bond acceptor. The comparative evaluation of these two distinct bioisosteres definitively establishes whether an N-H donor is an absolute requisite for robust SV2A target engagement.
In conclusion, the methodologies established in this study successfully detail the targeted structural evolution of the Levetiracetam scaffold. While the predicted physicochemical properties and structure-activity rationale hold significant promise, the ultimate validation of these four derivatives necessitates rigorous in vitro SV2A binding affinity assays and in vivo anticonvulsant screening (such as maximal electroshock and 6-Hz therapy-resistant models). The outcomes of these future pharmacological evaluations will provide critical validation for our structural hypotheses, ultimately advancing the rational design of potent, metabolically stable antiepileptic therapeutics.
CONCLUSION
Despite the availability of modern treatments, the epilepsy is still one of most of the common and difficult neurological disease in world. Almost a third of patients have seizures that will not go away. While Levetiracetam (LEV) has revolutionized epilepsy management through its unique modulation of the synaptic vesicle protein 2A (SV2A), its metabolic vulnerability specifically the rapid enzymatic hydrolysis of its primary acetamide group presents a critical pharmacokinetic limitation. This thesis successfully addressed this therapeutic challenge by conceptualizing, designing, and detailing the synthetic pathways for four novel LEV derivatives, systematically engineered to resist enzymatic degradation while optimizing target receptor engagement.
The structural modifications were strategically executed across three rational drug design paradigms: N-alkylation, extreme steric hindrance, and heterocyclic bioisosteric replacement. The N-methylated derivative was synthesized to evaluate the SV2A binding pocket's hydrogen-bond donor requirements while imparting mild steric protection. Pushing the steric boundaries further, the α-tert-butyl analog was designed to completely encapsulate the fragile amide bond within a massive hydrophobic shield, a modification that also holds the theoretical potential to reduce P-glycoprotein efflux and enhance central nervous system retention. Most notably, the development of the 1H-tetrazole and 1,2,4-oxadiazole bioisosteres provided a sophisticated chemical solution to completely bypass amide hydrolysis. These derivatives mimic the planar and electrostatic properties of the native carboxamide while conferring absolute resistance to type B esterases, allowing for a precise evaluation of the electronic interactions required for optimal SV2A binding.
In summary, this study offers a broad synthetic and theoretical basis for the development of the Levetiracetam scaffold. This work fully addresses the structural deficiencies of the parent drugs, yielding four promising options estimated to exhibit greater metabolic stability and improved blood-brain barrier permeability. To firmly validate their pharmacological profiles, extensive in vitro binding studies and in vivo anticonvulsant models are necessary; however, these new compounds provide essential insights into structure-activity relationships. In the end, this work is a big step forward in the rational creation of new, metabolically stable antiepileptic drugs that will help patients with drug-resistant epilepsy.
REFERENCES


Manmohan Kumar Nishad, Jitendra Kumar Yadav, Chemical Modification of Levetiracetam Acetamide Moiety to Enhance Anti-Convulsant Efficacy, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 4276-4307, https://doi.org/10.5281/zenodo.20731037
 10.5281/zenodo.20731037
10.5281/zenodo.20731037