
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Regulatory Affairs, Dayanand College of Pharmacy, Barshi Road, Latur, Maharashtra
The CTD offers a uniform framework for regulatory submissions that is acceptable in every ICH nation. Pharmaceutical items must submit a regulatory dossier through the CTD in order to receive marketing authorization. It defines the dossier as a collection of records outlining the effectiveness, safety, and quality of a product that needs to be turned in to regulatory bodies. The Common Technical Document (CTD), a widely used, standardized format that divides data into five modules, is the subject of the review. The Hatch - Waxman Act's unique submission process for generic medications in the US is called the Abbreviated New Drug Application (ANDA). Demonstrating therapeutic equivalency and bio-equivalence (BE) to the Reference Listed Drug (RLD) utilizing pharmacokinetic metrics like C_{max} and AUC is crucial to this approval. The legal environment is also covered, including the four kinds of patent certifications mandated by the ANDA. Citing the Central Drug Standard Control Organization (CDSCO) and its statutory duties under the governing Drugs and Cosmetics Act, 1940 and Rules, 1945, the report concludes by outlining India's regulatory structure. The CDSCO is overseen and managed by the Directorate General of Health Services, which is part of the Government of India's Ministry of Health and Family Welfare. Generic manufacturers are required to submit an Abbreviated New Drug Submission (ANDS). In the European Union (EU), medicinal items are subject to a distinct, intricate set of approval procedures and are heavily regulated.
A collection of documentation pertaining to the effectiveness, safety, and quality of a medical product is referred to as a dossier. A dossier submission is a collection of documents and information that summarizes the whole history of the creation and development of a product. The regulatory body must review and approve the dossier in order to authorize the product's marketing. The dossier includes all of the technical information on a pharmaceutical product, including administration details, information about the drug product's quality, safety, and efficacy, extra information and papers, samples, and reagents. Product chemistry, formulation, manufacturing, toxicity, pharmacology, pharmacokinetics, and clinical investigations are among the details included in the dossier. A product's marketing approval is contingent upon its dossier, which serves as a conduit for regulatory approval. The dossier is also referred to as a simple registration dossier, a pharmaceutical dossier in India, a marketing authorization application (MAA) in the EU, or a new drug application (NDA) in the USA. The Common Technical Document (CTD) is the standard format and structure that the dossier adheres to. The CTD, a standardized format (template) for data presentation in the ICH territories, is used to submit the dossier. There are five modules in the Common Technical Document. Administrative and regional data, which may vary by nation, are included in Module 1. All regions share Modules 2 through 5, which include summaries, quality, non-clinical, and clinical data, respectively.It is optional in several nations. Marketing authorization is the process of examining and evaluating a dossier to support a pharmaceutical product in light of its marketing (also known as license, registration, approval, etc.), which is obviously completed by granting a document. A dossier is a file that is submitted in accordance with a regulatory agency's requirements for a drug product's approval. In the USA and Europe, submitting a dossier file in the form of a common technical document is mandatory. Each nation has its own regulatory body that is in charge of enforcing laws and regulations and issuing guidelines to control medication marketing. Laws and regulations, such as the US Code of Federal Regulation (CFR) and the EU Directives, govern the dossier submission and review procedure. The general and permanent rules and regulations (also known as administrative law) that are published in the Federal Register by the executive departments and agencies of the US federal government are codified in the Code of Federal Regulation (CFR). The National Archives and Records Administration includes the Federal Register. The Office of the Federal Register and the Government Publishing Office (GPO) publish the general and permanent regulations in the CFR annual edition. The CFR is also available online in an unofficial format on the Electronic CFR website, which is updated every day, in addition to this annual edition. EU directives are a kind of EU legislation that requires member states to accomplish a particular objective while allowing them to select the means and format for doing so. Within a certain amount of time, usually two years, member states must incorporate the directive into their national laws. Generic medications authorized via MAA submission (EU) and ANDA submission (USA). After the original drug's patent expires, manufacturers of generic medications must demonstrate that their product is both therapeutically and bioequivalent to the original drug. A generic medication is designed to have the same dosage, safety, strength, and quality as a brand-name medication that is currently on the market. Patent-protected medications are known as brand medications. Approximately 47% of prescription medicine products in 2002 were generic, whereas 53% were innovator products. Due to patent expiration, there is approximately $4 billion in commercial potential for the next four years per year. Concerning difficulties were linked to the current development and approval of generic pharmaceutical goods.
In order to receive commercial approval, the safety, effectiveness, and therapeutic equivalency of such a medicine are evaluated early in comparison to the innovator or brand-name medication. However, in order to enable the production, approval, and marketing of generic pharmaceutical products, legislative, regulatory, and scientific difficulties still provide a barrier to the generic pharmaceutical business. Manufacturers of generic medications must create a product with the same clinical performance and therapeutic efficacy as their name-brand equivalent. ANDA submissions are used to approve generic medications. An application for a U.S. generic medicine approval for an already-approved or licensed medication is known as an Abbreviated New medicine Application (ANDA). The ANDA includes information that, when submitted to the FDA's Office of Generic Drugs, Center for Drug Evaluation and Research, allows for the evaluation and final approval of a generic drug product. After being accepted, an application may produce and sell the generic medication to offer the American public a low-cost, safe, and effective substitute.EU directives are legally binding agreements that mandate that member states accomplish a particular objective within a predetermined time frame, while they are free to select how to do so. Examples include Directive (EU) 2019/790 on copyright and related rights, which attempts to harmonize copyright laws across member states to support the internal market, and Directive on Administrative Cooperation (DAC7) (EU Council Directive 2021/514), which mandates that online marketplaces report seller data.
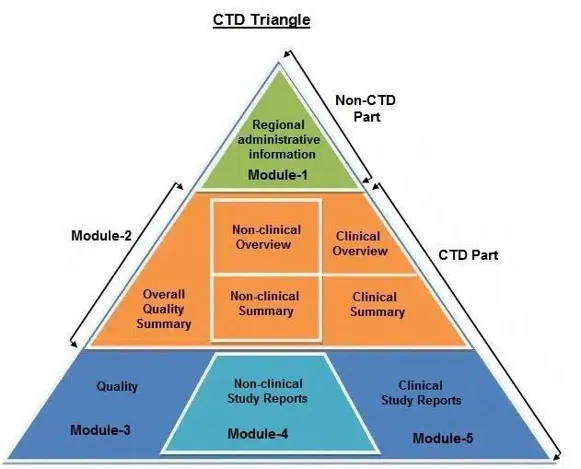
Fig 01: CTD Triangle
The International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) is a project that brings together experts from the pharmaceutical industry in the three regions as well as regulatory authorities from Europe, Japan, and the United States to discuss scientific and technical aspects of pharmaceutical product registration. By suggesting methods to create better harmonization in the interpretation and application of technical guidelines and requirements for product registration, ICH aims to minimize or eliminate the need to duplicate the testing conducted during the research and development of new medicines. In addition to eliminating needless delays in the global development and availability of new medicines while upholding quality, safety, and efficacy safeguards and regulatory obligations to protect public health, harmonization would result in a more economical use of human, animal, and material resources. In the pharmaceutical industry, a Reference Listed Drug (RLD) is a brand-name medication that has been approved by the FDA and is used as a standard for generic versions. To be approved through an ANDA, a generic medication manufacturer must demonstrate that its product is bio-equivalent to the RLD.
The RLD guarantees that generic medications are just as safe, potent, and high-quality as their original counterparts. By definition, an RLD is an FDA-approved medication that a generic medicine producer may use when submitting an ANDA (Abbreviated New medicine Application). In essence, an RLD is helpful in determining whether a product is bioequivalent to one that has already received approval. A generic manufacturer should cite the FDA-designated RLD in their ANDA application to demonstrate that the proposed generic medication is identical in terms of its active ingredients, dosage form, administration method, strength, labeling, and usage circumstances, among other features. The FDA's Approved Drug Product with Therapeutic Equivalency Evaluations, a book that includes all approved drug products approved based on safety and effectiveness, is most frequently referred to as the "Orange Book." It lists FDA-approved pharmaceutical drugs and offers details on their therapeutic equivalency. It assists researchers and medical professionals in finding safe and efficient generic medications that are comparable to name-brand medications. It contains information regarding patents and exclusivity as well as safety and efficacy data. It is regularly updated and accessible electronically as the electronic orange book.
The Hatch-Waxman Act is another name for the "Drug Price Competition and Patent Term Restoration Act of 1984." The Federal Food, Drug, and Cosmetic Act (FD&C Act), a 1984 US federal law that promotes the production of generic medications by the pharmaceutical industry and established the current system of government generic drug regulation in the US, was amended to create the approval pathway for generic drug products, under which applicants may submit an abbreviated new drug application (ANDA) under section 505(j). An act to support generic medications and innovators was required to address the aforementioned issue. The official act "The Drug Price Competition and Patent Term Restoration" was introduced by two American legislators, Orrin Grant Hatch and Henry Arnold Waxman, in 1984. Since then, it has been referred to as the Hatch-Waxman Act.
AIM: To make available more low cost generic drugs. The primary regulatory authority in India for pharmaceutical, medical device, and clinical trial regulation is the Central Drug Standard Control Organization (CDSCO). The Central Drug Authority for the discharge function is called CDSCO. assigned to the Central Government in accordance with the 1945 regulations and the Drug and Cosmetics Act of 1940. CDSCO's headquarters are in New Delhi. operating under the direction of the Ministry of Health and Family Welfare, Directorate General of Health Services, Government of India.
Vision: To safeguard and advance India's health.
Mission: Our mission is to ensure the safety, effectiveness, and quality of medications, cosmetics, and medical devices in order to protect and improve public health.
ANDA (Generic) drug Approval Process
When submitted to the FDA's Center for Drug Evaluation and Research, Office of Generic Drugs, an Abbreviated New Drug Application (ANDA) comprises information that allows a generic drug product to be reviewed and eventually approved. After being accepted, a candidate may produce and sell the generic medication to offer the American people a low-cost, safe, and effective substitute. The Drug Price Competition and Patent Term Restoration Act of 1984, also known as the Hatch-Waxman Act, established the Abbreviated New Drug Application (ANDA) system, which governs the complex, multi-stage regulatory and legal process of approving generic drugs in the United States. The goal of this system is to make less expensive generic medications more widely available while maintaining the same high standards of efficacy, safety, and quality as the original brand-name medication. The U.S. Food and Drug Administration (FDA), in particular the Center for Drug Evaluation and Research (CDER) through its Office of Generic Drugs (OGD), is in charge of the entire procedure.
The fundamental tenet of the generic approval procedure is that the brand-name drug's extensive and expensive safety and efficacy clinical trials do not need to be repeated for the generic. Rather, the generic manufacturer needs to demonstrate therapeutic equivalency and sameness to the FDA-approved brand-name medicine known as the Reference Listed Drug (RLD).
An ANDA must provide scientific proof that the suggested generic medication is comparable to the RLD in the following areas in order to be approved:
The scientific evidence that the generic medication functions in the body similarly to the RLD is known as bioequivalence (BE). It guarantees that the generic medication enters the bloodstream with the same quantity of active substance for the same amount of time.
The generic development process begins long before the application is submitted and involves intense scientific and legal preparation.
Reguanes Tompanyin Marange's book is the hit. One of four Patent Certifications is used for this.
|
Certification Type |
Meaning |
Legal/ Regulatory Outcome |
|
Paragraph I |
For the RLD, no patent information was submitted. |
No obstacle to approval (if all other requirements are fulfilled). |
|
Paragraph II |
The patents are no longer valid. |
There is no obstacle to approval. |
|
Paragraph III |
Until the patent expires, the generic will not be marketed. |
Until the patent expires, approval is postponed. |
|
Paragraph IV |
The generic medication won’t violate the patents or they are invalid or unenforceable. |
Requires the brand-name manufacturer and patent owner to be informed by the ANDA applicant frequently results in patent litigation. |
After the application dossier is finished, it is electronically submitted through the FDA's Electronic Submissions Gateway (ESG) as an Electronic Common Technical Document (eCTD). The Generic Drug User Fee Amendments (GDUFA) performance goals govern the review process.
Multiple professional scientific and regulatory teams concurrently review the filed ANDA:
The ANDA must be free of all patent and regulatory exclusivity obstacles prior to ultimate approval:
The Hatch-Waxman Act offers a sizable compensation to encourage generic businesses to contest dubious patents:
The Common Technical Document is divided into five modules:
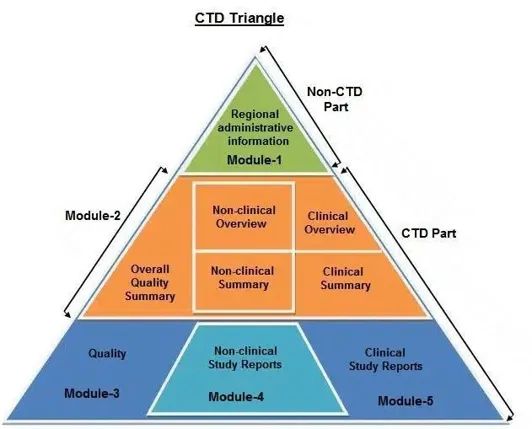
Fig. 02: CTD Triangle
Module 1 - Administrative & Prescribing Information
All administrative documentation (such as application forms, certifications, and claims of categorical exclusion) and labeling, including the documents listed below, should be included in Module 1. The following order should be used to arrange documents. Environmental evaluations should be submitted individually, but all of the materials in Module 1 can typically be included in one volume.
Form [kind of form]
1.11 Modification of information: Data not addressed in modules 2 through 5
Module 2- Summaries
Module 3 - Quality
3.2 Data body
3.3 References to literature
Module 4 - Nonclinical Study Reports
Module 5 - Clinical Study Reports
MARKETING AUTHORIZATION PROCEDURES:
Directive 2001/83/EC and Regulation (EC) No 726/2004 are the main regulations governing the strict, highly standardized procedure of submitting a drug product dossier for a Marketing Authorization Application (MAA) in the European Union (EU). The goal is to obtain commercialization authorization, which is only given following confirmation of the medication's efficacy, safety, and quality by an appropriate regulatory body. The electronic Common Technical Document (eCTD), a globally accepted format intended to standardize the arrangement of technical data, is the foundation of the submission. Either National Competent Authorities (NCAs) use the Decentralized (DCP), Mutual Recognition (MRP), or National Procedures to oversee the application process, or the European Medicines Agency (EMA) uses the Centralized Procedure (CP). The European Medicines Agency (EMA) and the Heads of Medicines Agencies (HMA) are principally in charge of the intricate and highly regulated process of submitting a drug product dossier for Marketing Authorization (MA) in the European Union (EU). The dossier, formally known as the Marketing Authorization Application (MAA), needs to be presented in an electronic format that is globally standardized.
Centralized refers to obtaining clearance from a single location. One central body has approved the medication, and this is applicable to the entire European Economic Area. This European Economic Area, or EEA for short, consists of the 27 EU member states as well as three other nations: Iceland, Norway, and Liechtenstein. Manufacturers can submit a single Marketing Authorization application (MAA) to the EMA thanks to the centralized process. Following the European Commission's approval, the centralized process, which is governed by Regulation (EC) 726/2004, permits manufacturers to promote their products to healthcare professionals. In this case, the European Commission is in charge of granting marketing authorizations. The primary benefit of this process is that, once marketing authorization is obtained, novel, cutting-edge pharmaceuticals can be made simultaneously accessible to all citizens of Europe. Because only one or two Member States are required to generate evaluation reports for any pharmaceutical product, the centralized process also results in increased efficiency throughout Europe.
Method:
Pharmaceutical businesses submit a dossier to the European Medicines Agency (EMA) if they want to adhere to the centralized method. The Committee for Medicinal Products for Human Use (CHMP), the EMA's medicines assessment committee, evaluates the dossier.In theory, the CHMP has 210 days to make a judgment. This time frame could be extended to provide the business a chance to respond to inquiries. Businesses are also able to explain their provided dossier verbally. The European Commission receives an opinion from the CHMP and uses it to make the final decision.
The CHMP's opinion is often accepted by the European Commission in every way. The package leaflet and the Summary of Product Characteristics (SmPC) are decided upon when a favorable decision has been reached. The result is an EPAR, or European Public Assessment Report. If the view is negative, details regarding the reasoning behind this judgment are provided. A European marketing authorization number is assigned to products that have received marketing authorization through the Centralized procedure. Certain items, such as biotechnology-based pharmaceuticals and novel pharmaceuticals meant to cure conditions including cancer, AIDS, neurological disorders, and diabetes, must go through the Centralized procedure. Companies are able to choose between centralized and national registration for other unique items. Under the Centralized method, the EMA accepts applications and manages the evaluation process. The European Commission receives an opinion from the CHMP and makes a legally binding decision. On the CHMP, each European Member State has one representative and an alternate. For every pharmaceutical product, the CHMP appoints two rapporteurs who keep an eye on the product for its entire life cycle. Members of CHMP function in their individual capacities. They serve as a link between national and European systems. The MEB receives reports from the Dutch CHMP members in the Netherlands.
The Timeline of the Step-by-Step Centralized Procedure and Important Steps:
The European Commission, the EMA, and the applicant company are all involved in the intricate, multi-phase CP procedure. The 210-day active review period is usually subject to "clock-stops" during which the applicant is required to respond to inquiries posed by the scientific committees of the EMA.
Phase Prior to Submission (Months 18 to 7): Participation and Readiness For procedural preparation and regulatory alignment, the presubmission stage is essential.
Request for Eligibility: The applicant must formally request and obtain confirmation from the EMA of the product's eligibility for the CP if it falls under the optional scope.
Intention to Submit & Pre-Submission Meeting: In order to ensure that the dossier is comprehensive and compliant, it is highly advised that the applicant notify the EMA of its intention to submit the ENA six to nine months prior to the scheduled submission.
Appointment of Rapporteur/Co-Rapporteur: The Rapporteur and Co-Rapporteur, two experts from two distinct EU/EEA Member States, are appointed by the EMA's Committee for Medicinal Products for Human Use (CHMP) to oversee the scientific evaluation.
MAA submission: The applicant uses the Electronic Common Technical Document
(eCTD) format to submit a single Marketing Authorization Application (MAA) dossier through the EMA's Submission Gateway. Q
Validation: The dossier is examined for administrative, legal, and technical
completeness by the EMA and the designated Rapporteur/Co-Rapporteur. The 210-day assessment clock begins on Day 1 if it is finished.
The CHMP is in charge of this crucial scientific review phase, which makes use of the knowledge of national regulatory bodies.
Primary Evaluation (Day 1 to Day 120): The Rapporteur and Co-Rapporteur create their initial assessment reports after thoroughly reviewing the clinical, non-clinical, and quality data. The suggested Risk Management Plan (RMP) is concurrently evaluated by the EMA's Pharmacovigilance Risk Assessment Committee (PRAC).
Around Day 120, the first clock-stop: The CHMP develops a thorough List of Questions (LoQ) for the applicant based on the preliminary reports. The clock is halted, usually for three to six months, so that the business can create and gather answers, update the product details, and add new data if needed.
Secondary Assessment (Days 121-180): After the answers are submitted, the clock restarts. After the submissions are reviewed by the Rapporteur and Co-Rapporteur, the CHMP may issue a second, shorter List of Outstanding Issues (LoOl), which could lead to another, shorter clock-stop.
A Final Assessment (Days 181-210): The CHMP completes its evaluation, develops its final scientific judgment on the medical product's benefit-risk balance, and examines the final product information materials (Summary of Product Characteristics (SmPC), Package Leaflet (PL), and labelling) in English.
4. EC Decision and CHMP Opinion (Day 210 onwards):
CHMP Opinion (Day 210): On the issuance of the marketing authorization, the CHMP adopts a final, unified opinion, either positive or negative. Positivity indicates that the advantages exceed the dangers.
Decision of the European Commission (roughly from Day 210 to Day 277): The European Commission receives the CHMP opinion and supporting documentation from the EMA.
After obtaining the CHMP view, the EC has 67 days to make the final, legally binding judgment. A single MA is now valid in every EU/EEA Member State thanks to the EC's ruling.
Publication of the EPAR: For each approved medication, the EMA publishes the European Public Assessment Report (EPAR), which is a comprehensive, publicly accessible scientific record of the evaluation.
Post-Authorization Activities:
The MA is renewable and has a five-year initial validity period. The MAH has ongoing responsibilities:
Pharmacovigilance: The MAH is required to keep an eye on the safety of the medication, report any adverse reactions, and submit Periodic Safety Update Reports (PSURs) to the EMA/PRAC.
Risk Management Plan (RMP): The MAH is also required to carry out and maintain the risk-monitoring and risk-mitigation activities specified in the approved RMP.
Variations: A variation application must be submitted to the EMA for any modifications to the MA, such as modifications to production, shelf life, or clinical data.
Renewal: Based on a reevaluation of the benefit-risk balance in light of post-marketing data, the MA must be renewed following the first five years.
For medications that have not yet received EU approval, decentralized process (DP) is applicable. Manufacturers may seek for simultaneous licensing for these medications in multiple EU member states. The process is governed by Directive 2004/27/EC. Any one-member state in the DP may decide to assess the application on its own. It will be difficult to determine which approach is ideal for your situation, even though all of them would result in compliance. A regulatory process inside the BU for the approval of specific pharmaceuticals for humans and animals is known as the European Union Decentralized Procedure. The process entails an evaluation and recommendation from a single EU member state (the reference member state), which is then sent to the European Commission for a final ruling that applies to all EU nations (from national marketing authorization to European marketing authorization). This process has two advantages over the previous mutual recognition process: it is more efficient and streamlined, and it permits early EU-wide marketing permission of specific pharmaceuticals.
It should be mentioned that not all pharmaceuticals are subject to the EU DCP, and that both Member States and businesses are free to choose not to participate. A regulatory route for the approval of certain pharmaceuticals for humans and animals in the European Union (EU) is the EU Decentralized Procedure (DCP). The Mutual Recognition Procedure (MRP) was replaced by the DCP, which was implemented to simplify and improve the efficiency of the EU-wide authorization of pharmaceuticals. The DCP does not apply to all pharmaceutical items and is optional for both businesses and EU member states. Early EU-wide marketing authorization of certain pharmaceuticals and a more adaptable and efficient instrument for pharmaceutical regulation than the centralized process are two benefits of the EU DCP.
For this reason, the EU DCP is thought to be a more effective and beneficial route for the approval of pharmaceuticals within the EU. The decentralized procedure is a European authorization process that is founded on the idea that the Reference Member State (RMS) should recognize the evaluation. When an applicant does not currently have a marketing authorization in any country, the decentralized method should be employed to get one in multiple Member States. The applicant asks for one nation to serve as the procedure's Reference Member State (RMS). The RMS distributes the initial draft assessment report after 70 days. The applicant and the Concerned Member States (CMS) may then provide feedback. The RMS circulates a second draft assessment report on the 120th day of the assessment process based on the company's responses to the questions asked by the RMS and other Concerned Member States (CMS), including comments on the SmPC, package leaflet, and labelling texts. During the next ninety days, there is also a mutual recognition procedure wherein other Member States typically accept the RMS's assessment, unless there are significant objections based on a potentially serious risk to public health.
The Coordination group for Mutual Recognition and Decentralized Procedures (CMDh) will also have additional discussions in such circumstances.
The procedure will be terminated negatively and no marketing authorization will be given if the RMS still has objections on day 210 that are thought to pose a substantial danger to public health. No discussion takes place in the CMDh in this instance.
Dutch translations of the SmPC, package leaflet, labeling texts, and mock-ups are submitted, and a national marketing license is granted once all participating Member States reach a favorable view on the items in the mutual recognition and decentralized procedure.
Application of DCP time slot:
You can utilize the MEB Planning tool to support time slot requests for decentralized procedure (DCP) applications or DCP line-extension procedures with the Netherlands as Reference Member State (RMS). The planning tool displays the number of time slots available for each department each month.
You can choose a time slot after launching the planning tool; a digital form will show up. The completed and signed Request for RMS in a Decentralized Procedure form can then be attached, along with your contact information.
You can refer to the organizational structure overview to determine which department should receive your request. In compliance with Chapter 2 of the Notice to Applicants, the RMS assigns the procedure number. The procedure number, NL/H/1234/xx/DC, is structured in accordance with the MRP procedure numbers with DC added. Later versions do not have DC. Since it is a nationwide registration, the product also receives an RVG number at the time of DCP application submission. You will receive an email confirming receipt of your submission. Within three weeks of submission, the MEB will let you know if the time slot has been assigned or not.
Removing an application from a CMS while the process is decentralized:
An application from one of the Concerned Member States (CMS) may be withdrawn at any point during the process.
An application is always discussed in the CMDh through the 60-day procedure if it is withdrawn in one or more Concerned Member States during the mutual recognition procedure due to a potential serious danger to public health. The process will be submitted to the CHMP if there is no agreement in the CMDh. The marketing authorization in the RMS may also be impacted by the outcomes of the CMDh/CHMP negotiations. A 60-day procedure in the CMDh or, if required, a referral to the CMHP will not result from withdrawing an application during the validation period or during the first part of the evaluation (days 0-119) of the decentralized procedure. The CMDh will always review requests that are withdrawn during the second phase of the evaluation (days 120- 210) due to "potential serious risks to public health" using the 60-day method. This process will be forwarded to the CHMP if an agreement cannot be achieved. It is not feasible to withdraw an application from the RMS. An application may only be withdrawn in each of the participating Member States.
The Methodical Decentralized Process:
The DCP adheres to a standardized, organized schedule. There are three primary phases to the 210-day active regulatory clock for scientific evaluation.
Consultation with RMS: It is highly advised that the applicant notify and agree with the selected RMS well in advance of the submission date (e.g., at least two months prior).
Submission: All designated CMSs and the RMS receive the same MAA dossier (in eCT format) at the same time.
Validation: The application is checked for administrative and legal completeness by the RMS and CMSs. The process begins on Day 1 if it is valid.
RMS Assessment (Day 1-Day 120): The RMS performs a thorough scientific assessment of the clinical, non-clinical, and quality data.
Draft Assessment Report: By Day 120, the RMS provides the CMSs and the applicant with the Draft Assessment Report (AR), which includes the suggested SmPC, PL, and labeling.
CMS Evaluation and Remarks (Days 121-150): The CMSs have 90 days (days 121-210) to examine the documentation from the RMS.
On the other hand, initial remarks are usually sent by Day 150 (or Day 30 of the 90-day term).
Based on the answers and conversation, the RMS completes the evaluation and product details. If all participating Member States agree to grant the MA, the RMS concludes the process.
An arbitration process is initiated if a CMS expresses concerns about a possible major danger to public health that cannot be addressed by dialogue:
Referral: The Coordination Group for Mutual Recognition and Decentralized Procedures, or CMDh, has been notified of the situation.
CMDh Decision: Within 60 days, the CMDh aims to come to an agreement. A binding conclusion for the participating Member States is produced if consensus is reached.
Referral to CHMP/EC (Last Resort): The European Medicines Agency's (EMA) Committee for Medicinal Products for Human Use (CHMP) is tasked with rendering a final conclusion in cases when the CMDh is unable to come to an agreement. Similar to the last stage of the Centralized Procedure, this opinion results in a legally binding decision by the European Commission (EC).
Each participating Member State has 30 days to issue its national Marketing Authorization following the successful completion of the process (with national consensus or a legally enforceable ruling from the EC/CMDh).
All participating nations use the same SmPC, PL, and labeling texts; however, the applicant is required to produce translations into the native languages.
A regulatory procedure in the European Union (EU) and European Economic Area (EEA) called the Mutual Recognition Procedure (MRP) is intended to extend a national Marketing Authorization (MA) for a pharmaceutical product to other EU/EEA Member States. It is based on the idea that the other participating nations should acknowledge each other's scientific evaluation and decision made by the first Member State.
For pharmaceuticals that have already obtained an MA in at least one EU/EEA nation but are not included by the mandatory scope of the Centralized Procedure (such as some biotech goods or orphan medications), the MRP is required even if the applicant wants to commercialize the product in other countries.
When a product is intended to be registered in more than one member state and has already been granted marketing permission in at least one member state at the time of application, the Mutual Recognition Procedure (MRP) is utilized. When a product has national marketing authority in at least one member state, we can use this approach. This approach is predicated on the notion that all of the EU's member states recognize one another. They employ adequately standardized standards to evaluate their implementation.
Therefore, the principle behind MRP is that a license that has been authorized in one member state should be recognised in another. The Reference Member State (RMS) is the nation in which the product has already received approval. Concerned Member States (CMS) are the newly added member states. The mutual recognition principle is at the heart of the MRP. This implies that the other Member States (the nations where the applicant now seeks authorization) are, in theory, required to acknowledge the evaluation of a medicinal product's quality, safety, and efficacy once a Member State (the original authorizing country) has granted a national MA.
This promotes the free flow of pharmaceuticals and avoids the duplication of scientific review throughout the EU. The medicinal product must already have a national marketing authorization in at least one EU/EEA member state, which serves as the Reference Member State (RMS), in order to use the MRP.
This Member State has already received the first national MA. In addition to serving as the procedural lead for the mutual recognition process, the RMS is in charge of revising its Assessment Report (AR). All other nations base their decisions on the MA awarded by the RMS.
The applicant is applying for a new MA in these EU/EEA member states. The proposed Summary of Product Characteristics (SmPC), the labeling and Package Leaflet (PL), and the RMS's AR are all reviewed by the CMSs, who either express their acceptance or voice concerns based on possible substantial risks to public health.
Representatives from the national competent authorities (NCAs) of each EU/EEA member state make up this coordinating body. In order to guarantee the harmonization of the MA throughout the participating nations, the CMDh's vital role is to settle disputes between the RMS and the CMSs.
The RMS has already granted a marketing license for the Mutual Recognition Procedure (MRP). Unless they object on the grounds of a potentially serious risk to public health, the RMS's assessment report serves as the foundation for asking the other Member States to mutually recognize the marketing authorization (including the Summary of Product Characteristics (SmPC), package leaflet, and labelling text). The Coordination group for Mutual Recognition and Decentralized Procedures (CMDh) will hold additional discussions in these circumstances. After the initial MRP is finished, a marketing authorization holder may use the MRP many times for the same marketing authorization in order for other Member States to grant a marketing authorization. The Repeat Use Procedure (RUP) is the name given to this process. This allows for the granting of a marketing authorization in one or more additional Member States that did not participate in the MRP or DCP. When submitting a national application, it is preferable to indicate that you intend to conduct a Mutual Recognition Procedure (MRP) with the Netherlands as RMS following the conclusion of the application. After the national application process is complete, a time slot for the MRP can be sought. For a product for which a marketing permission has previously been obtained, a time slot may also be requested.
The Methodical Approach to Mutual Recognition:
The principal recognition phase of the MRP, an organized process with a set timetable, lasts for ninety days.
Dossier Update: The applicant must make sure that the current dossier, which was authorized by the RMS,complies all applicable national and EU regulations.
RMS Request: The applicant formally requests that the RMS assign a process number (such as X/H/XXX/XX/MR) and amend its Assessment Report (AR) to reflect the current approved status.
CMS Notification: The applicant agrees on a start date (Day O) and notifies the selected CMSs of the upcoming submission.
The applicant simultaneously submits to all CMSS the same MAA dossier (in eCTD format), which includes the most recent Assessment Report from the RMS and the currently approved product information texts (SmPC, PL, and labeling) in English.
CMS Evaluation and Remarks (Day 1-Day 90):
The CMSs examine the product details and the RMS's Evaluation Report. In contrast to the Decentralized Procedure, the CMSs are not expected to examine all of the scientific data (quality, safety, and efficacy); instead, they are mainly looking for reasons that would support non-recognition due to a possible substantial risk to public health.
Rounds of Comments:
Consensus and Closure (Day 90): The RMS formally ends the process on Day 90 if all CMSS accept its evaluation and the suggested product information texts.
A CMS is promptly referred to the CMDh for resolution if, by Day 90, they oppose to the authorization due to a potential substantial risk to public health.
CMDh Discussion (60 Days): The CMDh has sixty days to review the unresolved issues. The goal of all participating Member States is to come to an agreement on the necessary actions (e.g., modifying product information or addressing particular safety concerns).
Binding Decision: The CMDh adopts a final stance that is binding on all participating Member States if a consensus is obtained.
Referral to CHMP/EC (Last Resort): The issue is sent to the EMA's Committee for Medicinal Products for Human Use (CHMP) for scientific arbitration if the CMDh is unable to come to an agreement within the allotted 60 days. The European Commission (EC) receives the CHMP's view and renders a final, legally binding ruling that is applicable to all Member States.
Translations: The applicant must provide the CMSs with high-quality translations of the final, agreed-upon SmPC, PL, and labeling texts within seven days of the procedure's conclusion (Day 90, or the final CMDh/EC judgment).
Granting of MA: Based on the harmonized texts, each CMS has 30 days from the end of the process to give its national Marketing Authorization.
The applicant may use the Repeat Use Procedure (RUP) to acquire authorization in other Member States that were not included in the first procedure following the successful completion of an MRP (or a Decentralized Procedure). In essence, the RUP is a new, simplified MRP in which the new CMSS recognize the already-approved MA.
SCHEDULE Y
The Drugs and Cosmetics Act of India contains a component called component Y that offers rules for conducting clinical trials of novel medications in India. Since its initial introduction in 1983, Schedule Y has undergone multiple revisions to conform to the most recent scientific and regulatory standards. The main goals of Schedule Y are to safeguard the rights and welfare of clinical trial participants and to guarantee the safety and effectiveness of new medications before they are authorized for use in India. Every facet of clinical trial conduct is covered by Schedule Y, including data management, ethical review, informed consent, research design, and reporting.
Schedule Y has several important provisions, such as:
Schedule Y is essential for guaranteeing the efficacy and safety of novel medications in India as well as safeguarding the welfare and rights of clinical trial participants. Schedule Y's regulations aid in ensuring that clinical trials for novel medications are carried out in India in a way that is both morally and scientifically sound.
DRUGS AND COSMETICS ACT, 1940 AND RULES, 1945:
The government's 1931 appointment of the pharmaceuticals Enquiry Committee resulted in a number of proposals for regulating the import, production, and distribution of pharmaceuticals. However, the government was hesitant to put these suggestions into practice. The government passed the Import of Drugs Bill in 1937 in response to the nation's rebellion, but it had nothing to do with the production, sale, or distribution of drugs. Lastly, on April 10, 1940, the Indian Legislature passed the medications and Cosmetics Act to regulate the import, production, distribution, and sale of medications and cosmetics.
The Indian Parliament made amendments to this Act in 1955 and again in 1960, 1962, 1964, 1972, 1982, 1986, 1995, 2008, 2017, 2018, and 2019. This Act provides licenses for the import, production, and distribution of pharmaceuticals and cosmetics. While state governments designate licensing authorities to oversee the production, distribution, and sale of pharmaceuticals and cosmetics, the federal government regulates their import.
There are five chapters in the Act:
Chapter I: Overview
Chapter II: Organizations in charge
Chapter III: Importing medications and cosmetics
Chapter IV: Producing, marketing, and distributing medications and cosmetics
Chapter IV A: Provisions relating to Ayurvedic, Siddha &Unani drugs
Chapter V: Other
The Act and Rules' main goals are:
Drugs: All medications used internally or externally by humans or animals, as well as any substances meant to be used for the diagnosis, treatment, mitigation, or prevention of any disease or disorder in humans or animals, including preparations applied to the human body to keep insects like mosquitoes away.
Cosmetic: Any substance that is meant to be rubbed, poured, sprinkled, sprayed, introduced into, or otherwise applied to the human body or any part of it in order to cleanse, beautify, promote attractiveness, or change the appearance; this includes any substance that is meant to be used as a component of cosmetics.
Misbranded medications include: (A) those that are so colored, coated, powdered, or polished that damage is hidden; (B) those that are not properly labeled; or (C) those that are made to look better or more therapeutically valuable than they actually are.
Adulterated drug: A drug is considered adulterated if it contains, in whole or in part, any filthy, putrid, or decomposed substance; if it has been prepared, packed, or stored in an unhygienic manner, which could have contaminated it with filth or made it harmful to health; or if its container contains, in whole or in part, any poisonous or deleterious substance that could make the contents harmful to health.
Spurious drugs are: (A) imported under a name that belongs to another drug; or (B) imitating, substituting, or resembling another drug in a way that could lead to deception, or bearing the name of another drug on it, its label, or its container.
Manufacturer: Manufacture refers to any method or portion of a process for creating, modifying, decorating, completing, packing, labeling, disassembling, or otherwise handling or adopting any medication or cosmetic with the intention of selling or distributing it.
Schedules to the Acts:
Schedules to the Rules:
It outlines many forms that must be submitted in accordance with the Drugs and Cosmetics Act in order to submit applications for licenses, transmit memos, etc.
It specifies the costs the Central Drugs Laboratory and Government Analyst will charge for testing or analyzing drug samples.
The list of biological and other special items is prescribed in Schedule C&C (i).
It lists drug classes that are not subject to some of the rules governing drug imports.
A list of toxic compounds is prescribed, but it is not included (22/6/1982).
A list of Ayurvedic, Siddha, and Unani toxic substances is prescribed in Schedule E (i).
It lays out rules pertaining to blood bank regulations and obtaining a license to process blood components.
Schedule F(i) outlines regulations that apply to the manufacturing of sera, diagnostic antigens, and bacterial and viral vaccinations.
It establishes the requirements for surgical dressings.
Schedule F(iii) outlines the requirements for umbilical tapes.
It establishes the requirements for ophthalmic preparations.
It provides a list of medications that must only be administered under a registered medical practitioner's supervision. It has the following label: 'Schedule G Drug's Warning "It is dangerous to take this preparation except under the supervision of Registered Medical Practitioner"
It prescribes a list of medications that can only be sold in stores with a registered medical practitioner's prescription. The label for Schedule H medications states, "To be sold by retail only on the prescription of Registered Medical Practitioner."
In some situations, it calls for calculating the proportion of toxins; nevertheless, this was left out (22/6/1982).
It lists the conditions or illnesses that medications may not be able to prevent or treat.
Schedule K lists drug classes that are not subject to certain manufacturing-related regulations.
The list of medications that can only be purchased with a prescription is not included (22/6/1982).
It specifies the requirements for factory facilities, machinery, equipment, etc., as well as good manufacturing practices (GMP) for the production of pharmaceuticals.
Schedule M (i) outlines the specifications for industrial space, machinery, and other items needed to produce homoeopathic medications.
It outlines specifications for industrial space, machinery, and other items needed to produce cosmetics.
Schedule M (iii) outlines the specifications for industrial space, machinery, and other items needed to produce medical devices.
It specifies the minimal equipment that pharmacies must have.
Schedule O outlines regulations that apply to black disinfectant fluids.
It prescribes medications for a lifetime.
Schedule Pi) specifies the medication pack sizes.
It specifies the list of colors that can be used in coal tar cosmetics and soaps.
It specifies requirements for single-use rubber latex condoms.
Schedule R (i) specifies requirements tor medical equipment.
It establishes requirements for cosmetics.
For the production of Ayurvedic, Siddha, and Unani drugs, it specifies the standards for industrial space, machinery, equipment, and hygienic conditions
It specifies the details that must be displayed in medication production records.
Schedule U (i) specifies the details that must be included in cosmetics production records.
It establishes guidelines for private and patent medications.
It prescribes a list of medications that are only sold under generic names.
It lists a number of narcotic medications and psychotropic chemicals that might cause addiction and that require a license to import, manufacture, distribute, and sell.
It establishes standards and guidelines for clinical studies in order to import and produce new medications.
In essence, the Act (1940) establishes the "what" (definitions, objectives, prohibitions, and penalties) and the Rules (1945) establish the "how" (forms, procedures, standards, and specific requirements under the various Schedules) necessary for the effective regulation of the pharmaceutical and cosmetic industry in India.
CENTAL DRUG STANDARD CONTROL ORGANIZATION (CDSCO)
The National Regulatory Authority (NRA) of India is the Central Drugs Standard Control Organization (CDSCO), which is part of the Directorate General of Health Services, Ministry of Health & Family Welfare, Government of India. In addition to having six zonal offices, seven sub zonal offices, thirteen port offices, and seven laboratories dispersed throughout the nation, its headquarters is situated at FDA Bhawan, Kotla Road, New Delhi 110002. Central and state regulators have been given a number of tasks for the regulation of pharmaceuticals and cosmetics by the pharmaceuticals & Cosmetics Act of 1940 and the regulations of 1945.It envisions consistent application of the Act's provisions and the rules enacted thereunder to protect patients' rights, safety, and well-being by controlling medications and cosmetics.
To guarantee the safety, effectiveness, and quality of the medical products produced, imported, and disseminated in the nation, CDSCO continuously strives to increase accountability, transparency, and consistency in its services.In accordance with the Drugs and Cosmetics Act, CDSCO is in charge of approving new drugs, conducting clinical trials, establishing drug standards, regulating the quality of imported drugs in the nation, and coordinating the operations of State Drug Control Organizations by offering professional advice with the goal of bringing about uniformity in the enforcement of the Act. Additionally, CDSCO and state regulators work together to provide licenses for specific vital drug categories, like blood and blood products. V. Sera, vaccines, and fluids. The Ministry of Health and Family Welfare is home to the Drug Controller General of India (DCGI), who oversees pharmaceutical and medical device regulation within the CDSCO. The Drug Consultative Committee (DCC) and the Drug Technical Advisory Board (DTAB) provide advice to the DCGI.
Each of the zonal offices conducts post-market monitoring, pre-licensing and post-licensing inspections, and, if required, medication recalls. Manufacturers having the necessary authority must designate an Authorized Indian Representative (AIR) to act on their behalf in all interactions with the CDSCO in India.
Zonal Offices: Ghaziabad (North Zone), Kolkata (East Zone), Mumbai (West Zone), Chennai (South Zone), Ahmedabad (Zonal Ottice), and Hyderabad (Zonal Office) are the locations of the zonal offices.
They participate in GMP audits and inspections of large-scale parenteral, serum, vaccine, and blood product manufacturing facilities.
Sub Zonal Offices: Bangalore, Varanasi, Goa, Jammu, Indore, Guwahati, and Baddi are the locations of sub-zonal offices.
For standardized standards of inspection and enforcement, these centers collaborate with State Drug Control Authorities under their purview.
Port/ Air port offices: Delhi, Chennai (air and sea port), Kolkata (air and sea port, Mumbai (air and sea port), Cochin, Indore, Hyderabad, Vishakhapatnam, Krishnapatnam, and Ahmedabad are the locations of port/air port offices.
Laboratories
Responsibilities & Functions of CDSCO:
GENERAL CONSIDERATION FOR DOSSIER PREPARATION:
The CTD is merely a format for sending data to CDSCO.
The greatest guidelines for creating an acceptable document are common sense and a clear focus on the demands of the regulatory authority assessor, even though adherence to the general CTD structure is required. However, it should be emphasized that no guideline can cover every scenario. In order to communicate the information as effectively as feasible and to aid in comprehension and assessment, candidates may alter the format at certain of the subsection levels. The left-hand margin should be broad enough to prevent information from being covered by the binding method, and text and tables should be created with margins that allow the page to be printed clearly without losing any information. Text and table fonts should be large enough in both style and size to be easily readable.
It is advised to use a Times New Roman font of 12 points for descriptive text and 9 to 10 points for text and table items. Every acronym should be mentioned at the end of the dossier and described the first time it is used.
The most recent version of the uniform guidelines for papers submitted to biomedical journals should be followed for citing references. ICMJE stands for International Committee of Medical Journal Editors.
DIFFERENCE BETWEEN USA, EU & INDIA:
Table 1: Principle differences between US, EU & INDIA
|
Requirements |
USA |
EU |
INDIA |
|
Agency |
USFDA |
EMEA CHMP National Health Agencies |
DCGI |
|
Registration Procedure |
A Single Registration Procedure |
Mutual Recognition, Centralized, Decentralized & National Process |
A Single Registration Procedure |
|
TSE/ BSE Study data |
TSE/ BSE Study data is not necessary. |
TSE/ BSE Study data is necessary. |
TSE/ BSE Study data is necessary. |
|
Post-approval changes |
Changes made to an approved medication after approval: Minor changes Moderate changes Major changes |
Variation of the authorized medication: Variation in Type IA Variation in Type IB Variation in Type II |
Post-approval changes: Major quality changes Moderate quality changes |
Table 2: Administrative Requirements
|
Requirements |
USA |
EU |
INDIA |
|
Application |
ANDA |
MAA |
MAA |
|
Debarment Certification |
Needed |
NA |
NA |
|
No. of Copies |
3 |
1 |
1 |
|
Approval period |
18 Months |
12 Months |
12 Months |
|
Fees |
No Fees |
10-12 Lakh |
50,000 |
|
Presentation |
eCTD & Paper |
eCTD |
Paper |
|
Justification |
ICH Q6A |
ICH Q6A |
ICH Q6A |
Table 3: Finished Product Control Requirements
|
Requirements |
USA |
EU |
INDIA |
|
Assay |
90-100% |
95-105% |
90-110% |
|
Disintegration |
Not necessary |
Necessary |
Necessary |
|
Colour Identification |
Not needed |
Needed |
Needed |
|
Water Content |
Needed |
Not Needed |
Needed |
Table 4: Manufacturing & Control Requirements
|
Requirements |
USA |
EU |
INDIA |
|
No. of Batches |
1 |
3 |
1 |
|
Packaging |
Minimum 1,00,000 units |
Not needed |
Not needed |
|
Process Validation |
Not necessary at the submission time |
Needed |
Needed |
|
Batch size |
Minimum 1,00,000 units |
Minimum 1,00,000 units |
Not specified |
Table 5: Stability Requirements
|
Requirements |
USA |
EU |
INDIA |
|
No. of Batches |
1 |
2 |
1 |
|
Condition |
25/60; 40/75 |
25/60; 40/75 |
30/35; 30/70 |
|
Date & Time submission |
3 Months Accelerate & 3 Months Long Term |
6 Months Accelerate & 6 Months Long Term |
6 Months Accelerate & 3 Months Long Term |
|
Orientation of container |
Upright & Inverted |
Avoid addressing |
Avoid addressing |
|
Clause |
21 CFR Part 210 & 211 |
Volume 4, EU guidelines for medicinal product |
ICH Q1F |
|
QP Certification |
Not necessary |
Required |
Required |
Table 6: Bio-equivalence Requirements
|
Requirements |
USA |
EU |
INDIA |
|
CRO (Audit) |
FDA Audit |
MHRA Audit |
CDSCO |
|
Reserve Sample |
5 times the sample necessary for analysis |
No such requirements |
- |
|
Fasted/ Fed |
Must follow OGD recommendations |
No such requirements |
CDSCO recommendations |
|
Retention samples |
5 years from the application filing date |
No such obligations |
3 years from the application filing date |
RESULTS & CONCLUSION:
INDIAN MARKET:
The multi-step, quality-driven CDSCO (Central Drugs Standard Control Organization) process for obtaining product approval (dossier) for existing molecules is aimed at guaranteeing the product's efficacy, safety, and consistent quality manufacturing. The manufacturer submits a thorough dossier to start the application procedure. The application form, the whole manufacturing formula, and a synopsis of the manufacturing process are all included in this. A minimum of three months' worth of accelerated stability study data, a thorough testing process with specifications, and the proposed product label details are essential quality documentation requirements. The company must also give information on the technical experts in charge of the production and testing processes. The drug control department's pre-approval examination is a crucial stage. The purpose of this inspection is to confirm that the manufacturing facility complies with Current Good Manufacturing Practices (cGMP), particularly with regard to the requirements of the Revised Schedule M of the Drugs and Cosmetics Rules. The firm must also show that they have a sufficient infrastructure for quality control. The facility must either sign a contract with a certified commercial testing lab or have its own testing unit that specifically includes a microbiology division. Lastly, each commercial batch needs to be thoroughly tested before distribution in order to guarantee product quality throughout its life-cycle. If the product is official, this testing must meet the requirements of the Indian Pharmacopoeia (IP) or the company's internal specifications.
USA MARKET:
US Food and medicine Administration (USDA) standards for filing an Abbreviated New Drug Application (ANDA) for generic medicine approval. The manufacturer's dossier must include thorough information about product development in order for a generic medication to be approved through the ANDA process. This covers information on stability research, analytical development, and formulation development. The company must prepare important documents, such as a Master Batch Record, a process validation plan, comprehensive testing procedures with specifications, and a stability protocol, before starting the submission batch manufacture. The manufactured submission batch is important since it needs to be put through stability studies at the same time as a bio-equivalency (BE) study against the Reference Listed Drug (RLD). To verify control and consistency, a stratified sampling procedure must be used to validate the manufacturing process for this batch of submissions.
All these technical documents, along with the anticipated batch record-specifying the commercial batch size and the equipment to be used (particularly if different from the submission batch equipment)—must be included in the dossier. The filing firm must also establish a local office in the USA to serve as a liaison with the FDA as a procedural requirement. This local US office is the conduit for all official FDA correspondence pertaining to the ANDA or sANDA (supplemental ANDA).
EU MARKET:
For generic product marketing permission (dossier approval) in Europe, contact the European Medicines Agency (EMA). A thorough dossier covering quality, safety, and efficacy must be prepared and submitted to the European Medicines Agency (EMA) in order for a generic pharmaceutical to receive marketing authorization in Europe. Documentation Prior to Batch: The company needs to complete a number of foundational paperwork before starting manufacturing. These comprise the Master Batch Record, the process validation protocol, the stability protocol, precise testing procedures with specifications, and comprehensive product development information (formulation, analytical techniques, and stability studies). Submission Batch Manufacturing & Testing: To be used as exhibit/submission batches, at least two batches must be produced at the full commercial batch size. These batches are crucial because they need to be put through stability studies and a bioequivalency (BE) analysis against the reference product gathered from the market. For these particular batches, the production procedure needs to be thoroughly verified. Additionally, the principal packing materials and raw materials must be certified free of Bovine Spongiform Encephalopathy (BSE) and Transmissible Spongiform Encephalopathy (TSE) in order to reduce hazards. Regulatory and Post-Approval Requirements: Until the brand patent expires, the product cannot be approved. For the proposed product, the firm must submit at least 12 months of real-time stability research results to establish a 24-month shelf life. Importantly, in order to handle all regulatory communications, the company needs to establish a registered office that operates full-time in Europe. Should the firm desire to scale up the batch size during commercial supply, this modification must be legally approved by the EMA by submitting a variation application.
REFERENCES

Sakshi Swami, Nirbhay Chalmale, Dr. Kranti Satpute, Anjali Dudhate, Pratima Shinde, Comparative Study of Dossier Compilation & Submission Process of Drug Product in USA, Europe and India, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 4416-4458. https://doi.org/10.5281/zenodo.20736519
 10.5281/zenodo.20736519
10.5281/zenodo.20736519