
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
IPS Academy College of Pharmacy, Rajendra Nagar, A.B. Road, Indore–452012, (M.P) India.
Drug Discovery and Development are complex and expensive processes with time-consuming aspects. There are many reasons for the failure of drug discovery and development, including lack of efficacy, toxicity, and poor pharmacological properties of potential drugs. Computer-Aided Drug Design (CADD) and In Silico methods have become more popular in recent years, offering scientists a better alternative than traditional experimental methods. These new technologies incorporate computer-based modeling with bioinformatics and cheminformatics to improve the effectiveness of the drug discovery and development process in a shorter amount of time. As such, this review focuses on the major in silico drug discovery strategies, such as Target Discovery, Ligand-Based vs Structure-Based Drug Design, Molecular Docking, Virtual High-Throughput Screens (vHTHps), Quantitative Structure-Activity Relationship Analysis (QSAR), and ADMET Prediction. It will also explain how bioinformatics and Molecular Modeling help identify and optimize potential leads for drug discovery, along with describing the latest advancements in Artificial Intelligence and Machine Learning that are helping improve the overall process. Finally, this review will discuss the common challenges, limitations, and future directions related to computational drug design. Ultimately, utilizing in silico approaches will save time, save money, and reduce the overall costs associated with drug discovery and development, making them indispensable parts of the modern drug discovery and development landscape
The right amount of safety, pharmacokinetics (how often drugs will be absorbed into the body), and how a drug behaves (biochemistry) all play an important role in determining if that drug will work. If a drug does not have the right amount of Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMET) characteristics, it will not be as successful as a drug with high potency and selectivity. The drug must also be able to perform all functions well; that is, it must be well absorbed into your body, distributed to all tissues/organs, metabolized (digested) without losing its activity immediately after it enters the body, excreted from the body in a safe manner and have no toxicity associated with it. Although these factors may seem to be independent of each other in drug development, each factor impacts on the others and they are related (1). Due to this, computer-assisted drug design (CADD) methods have become more prevalent recently. CADD can help address size, timing, and cost issues associated with traditional laboratory methods used to develop drugs. CADD consists of using computers to identify drug targets, virtually screen large amounts of chemical libraries, further optimize selected compounds, and evaluate probable toxicity of these compounds in vitro and in vivo (via laboratory and animal testing). Following completion of all steps in CADD, laboratory and animal studies are then performed on drug candidates to verify that the computer predictions were correct. CADD improves success rates and reduces the number of chemical failures in drug development. (2). Because in silico techniques save time, cost and eliminate the requirement of performing lengthy in vivo studies, these methodologies have developed into critical tools for contemporary drug discovery and development. In addition to improving safety assessments, the use of these computer methods provides enhanced ADME profiles and the ability to forecast drug behaviour. Therefore, these techniques play an essential role in future drug development strategies. Although there exist limitations associated with the implementation of these technologies alone, they have contributed greatly to drug research by integrating them with pharmacology, toxicology and biotechnology (3).
1.1 Drug Discovery Using Traditional Methods
The traditional method of screening natural compounds for drugs was based on the empirical method, which took a long time to produce results and was often expensive, labour-intensive and produced random results instead of systematic results. The methodologies used to validate drug activity prior to sustainable use were not able to effectively consider important factors relating to the pharmacokinetics or bioavailability of the drug, nor did they adequately describe how the drug would interact with the intended target. However, the advent of genomics, high-throughput screening, structural biology and computational biology significantly transformed drug discovery from the use of traditional (old) and new (e.g., genome sequence) data, thus leading to improvements in the research of drug discovery. (4)
1.1 Current Trends & Challenges in Computational Optimization
To optimize the process of drug development, we must convert design goals into mathematical functions. This often creates complex problems that have multiple objectives. Recent popularity of nature-inspired metaheuristic algorithms, such as particle swarm optimisation, cuckoo search and firefly optimisation, can be attributed to their ease of use, Flexibility and ability to effectively solve Non-Linear problems. Use of surrogate models, while reducing costs of computation can lack consistency in rules, and often involve trade-offs with regard to accuracy. Given that they do work, there are still significant issues associated with broadscale implementation of many different types of meta-heuristics in practice and a lack of reliable theoretical evidence supporting that they converge (5).
1.1 Definition and Scope of In-Silico
"In-silico" means using computers to run simulations that can predict how substances will behave. Both the EU and EPA define "in-silico" as data models that require no further testing, however, the EU considers it a data model based primarily on the assumption of no testing based upon computer generated data while the EPA considers it an amalgamation of computing plus molecular biology. The use of in-silico methods, for example, to predict biological activity, efficacy, and toxicity will reduce animal testing, optimise drug distributions and expedite drug manufacture and development. The uncertainty surrounding the length of time required and the amount of expense involved with in vitro and in vivo methodologies has resulted in an increasing use of in-silico technologies; notwithstanding the fact that these traditional methodologies (both in vitro and in vivo) are required by law in most jurisdictions, they also present ethical concerns regarding the act of using animals. The reliability of in-silico methods relies on the databank of information available, and requires both external validation and a systematic review of the methodology being used prior to their use; thus, for a reliable and/or valid prediction of pharmacology/toxicology from in-silico methods, the methodology must be valid, sensitive, specific, and at least take into account the limitations of the dataker and/or the intended use of the predictability (3).
1.1 Objectives of the Review:
• To explain in detail what computer aided drug design (Cadd) is and how it is used in modern drug development.
• To talk about how Cadd assists with hit detection, hit-to-lead selection, and ADMET property optimization without putting anyone in danger.
• To point out the problems with traditional methods like high-throughput screening (Hts) and combinatorial chemistry when it comes to finding certain biological targets.
• To explain how in silico methods can speed up and lower the cost of finding lead compounds.
• To show how Cadd is utilized in every step of drug research, from finding targets to preclinical and clinical trials.
• To show how adopting in silico technology in research and development can save the time and expense of making drugs by up to 50% (6).
(i) There are many factors that influence the way drugs are developed and discovered. The most significant factors are described below: Therapeutic Objective: The therapeutic objective exists as a limit to the development of therapeutic drugs. For example, an antacid is easier to develop than a specific proton pump inhibitor. The therapeutic objective determines the options for finding new drugs that work or do not work. Skills of the Medicinal Chemists: The skills of the chemist in understanding the biology of the disease state and in understanding the chemistry of the compound used as the lead molecule will influence the ability to develop new medicinal products. Screening Facilities: The primary consideration for an effective quick method for performing high throughput screening is the ability to conduct evaluations of large quantities of compounds and identifying those compounds that may produce favorable responses in a clinical setting. Drug Development Facility: In order to develop drugs it is important to have facilities that provide an environment for the collaboration of individuals from various disciplines such as medicine, chemistry, biology, and pharmacy. The three factors that will influence the cost of drug development are (7).
(iii) Synthesis of compounds (chemicals): From the 5000-10000 tested compounds only 1 drug will be released for general use.
(iv) Lead molecule characteristics: The development cost of a lead molecule will be extremely high if an expensive manufacturing process is used.
(v) New drug development requirements: Prior to this procedure the developmental cost requirement based on costs is approx. 33% of what it currently is, the amount of time spent developing something used to take 10 to 16 years now will be 6 to 8 years only.
The process of in silico drug discovery consists of three stages:
1) Identification of the therapeutical target, and development of multiple small compound libraries to evaluate therapeutical lead candidates, followed up with development of virtual screening methods such as identifying potential small molecule binding sites using existing molecules from a compound library or use of de novo design methods to create small molecules in the target binding site.
2) Evaluate the specificity of the lead compound candidates using binding site information available for other known pharmaceutical compounds
3) Conduct enhanced in silico ADMET profiling evaluations of the lead candidates and assign lead candidate status to any compounds that pass the criteria established during the profiling evaluation process.
2. CORE METHODOLOGIES IN COMPUTER-AIDED DRUG DESIGN (CADD)
The next part talks about several important methods utilized in research on in-silico drug design.
2.1 Homology modeling
Sometimes referred to as comparative protein modeling, homology modeling uses the experimentally-determined 3-dimensional (3D) structure of a homologous protein to create a model of the 3D structure. The 3D structure of the homologous protein, referred to as the 'template', serves as the basis for generating a model for the hypothetical 3D structure of a protein with an unknown atomic resolution based on the amino acid sequence of its target. This model generation method is called homology modeling. The first step in using homology modeling is to identify one or more representative protein structures similar to the query protein sequence and align the corresponding template and query protein residues. By aligning the corresponding residues of the template protein and query protein, and using the multiple sequence alignments of the respective representative protein structures, it has been demonstrated that, while homologous protein sequences may have considerable sequence differences and have less than 20% sequence identity, the corresponding 3D structures of proteins tend to have more conserved structural features than do homologous protein sequence features291, 321 (8,9,10).This indicates that homologs of evolutionarily related proteins maintain identical alignments of equivalent protein residues with homologous sequences, regardless of whether they are biologically-produced from natural gene products or synthetically-produced from man-made sources319(6,9).
2.2 Structured based drug design (SBDD)
SBDD represents one of the earliest methods for creating medicines. Generally speaking, the therapeutic targets of a drug are the biological molecules involved in cellular signalling or metabolism that are believed to be associated with a disease state. Therapeutical targets are often proteins, and enzymes involved in the aforementioned pathways. The pharmacological action of a drug is to inhibit, revert or modify the structure and/or function of a disease-causing protein/enzymes. SBDD creates new drugs using structural information available to researchers on a three-dimensional basis (i.e., using the actual shape or structure of a target protein). Techniques used to obtain three-dimensional structures of target proteins include nuclear magnetic resonance and x-ray crystallography. Structures of proteins are determined using nuclear magnetic resonance and x-ray crystallography; resolutions of both methods are in the range of angstroms. The distance of one angstrom is equal to approximately 1/100 of the width of a human hair, which is 500,000 times smaller than the cut section of a human hair. At such high resolutions, it is possible for researchers to investigate how the atoms within proteins interact with the atoms of pharmacologically active molecules. Therefore, SBDD is a successful method of designing drugs and can provide high resolution structural information for both proteins and small molecules (i.e., CCBDs). SBDD is successful in assisting the match up of the aforementioned. (10).
2.3 Target identification
The function of a potential drug target, which has a connection with an illness, is the initial step of target-based drug development. Since a target will normally be defined as a single molecule, either a gene or a protein; therefore, it will be defined as having an association with that particular illness. A therapeutic target can be defined as a necessary molecule within a specific metabolic or signalling pathway associated with a disease or pathology; or an ability of a microbial pathogen to grow/proliferate or survive. The following are examples of strategies when someone has an illness: some strategies want to block the function of an important molecule so that the pathway is non-functional; and/or to develop drugs which bind to the active site to block the defined molecule from functioning. Another strategy is to stimulate the appropriate "molecules" to enhance the normal pathway if it was damaged during the course of the illness. Determining the specific target and related patient population is accomplished by methodologies utilizing bioinformatics, chemo-informatics, and/or data mining methodologies. This includes but is not limited to: homology-based, ligand-based, structure-based, high-throughput screening (HTS), text mining, microarray technologies, and pattern matching. (10).
2.4 Ligand-based drug screening
LBDD, or ligand-based drug design, relies on existing information to make projections for new molecules that may have the same biological effects. Thus, when projecting drug molecules the basis for this projection is that the more similar two compounds are in their structural relationships as well as their physicochemical characteristics (eg., molecular weight, hydrophobicity, number of anionic and cationic residues and number of hydrogen bond donors and acceptors, aromaticity, etc.), the greater the potential for the existence of similar biological activities. This is accomplished through the use of a comparative analysis of different characteristics surrounded by each chemical compound. LBDD is also applied when the three-dimensional structure of the protein of interest is not known. When the protein's structure is not known, both QSAR and pharmacophore modeling methods can provide excellent information about how the ligands will bind to and interact with the target protein.
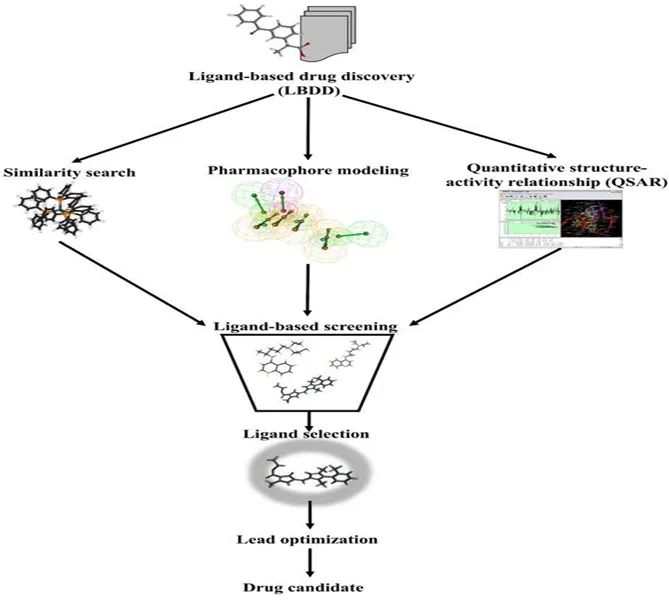
Fig. 1: Schematic representation of ligand-based drug discovery and screening approaches
2.5 Quantitative SAR
The QSAR method is established on the premise that similar molecules should, in general, have likewise biological actions. Mathematical models, also called "QSAR models," provide a way to predict how the binding of a ligand affects the interactions between ligands and the appropriate target molecule based on how the binding of different types of ligands to the target molecule leads to the expected biological actions resulting from that interaction. That is, by evaluating how the different structural components of a ligand that bind to its target are associated with lab-based biological activity, it is possible to create estimates of the biological activity of similar new ligands. Therefore, the predictive ability of QSAR models is solely based on the applicability of the chemical descriptors used for predicting the biological activity of the ligands that are chemistries represented by the QSAR model. Further, once the "hit" is identified in a high-throughput screen or database query, the QSAR model should describe those ligands that bound to the target molecule and provide a description of the biological action that might be associated with each ligand. The molecular structure and/or chemical composition of an organism informs the functions of an organism, including the number of atoms within each molecule and/or multiple molecules; the number of individual bonds present within each molecule or between molecules; the total surface area represented by each molecule; and the functional groups associated with each molecule. Therefore, after determining the structural and/or physicochemical properties of a set of pharmaceutical compounds, a QSAR model can be developed that documents the effects of those properties on living organisms. Subsequently, the QSAR model can be used to predict the potential for improving the performance of active pharmaceutical ingredients in vivo. Ultimately, researchers will provide the results of their studies to demonstrate that a new active agent can demonstrate an increased potential for improving or promoting health. (3).
2.6 Molecular docking
Molecular docking studies how small molecules interact with target protein binding sites, and as more protein structures are identified through nuclear magnetic resonance (NMR) and X-ray crystallography, molecular docking has become more widely used in drug discovery. It can now be done by using molecular docking techniques to determine the binding potential of previously unknown protein structures to predicted binding targets based on homology. The use of docking methods to evaluate the drug-like properties of various compounds and their compatibility with specific targets will support lead optimization. Molecular docking software evaluates the shape of ligands repeatedly until finding an equivalent low-energy conformation. To rank all potential conformations following this procedure, affinity scoring functions are used; this is accomplished by calculating the electrostatic and van der Waals contributions to the total binding energy from each conformation, resulting in a total ΔG value (in kcal/mole). In biological systems, the forces that drive interactions among ligands or substrates and corresponding binding-site surfaces maximize complimentarity of shape and electrostatics, thus providing comprehensive strategies for the identification of potential drug candidates and assessing those candidates relative to their corresponding targets in drug design. (3).
3. KEY TECHNIQUES AND TOOLS IN THE IN-SILICO PIPELINE
3.1 High-Throughput Virtual Screening (vHTS)
The pharmaceutical industry has a long history of attempting to create innovative drugs. Virtual High Throughput Screening (vHTS) provides the means to achieve this objective. vHTS uses protein target molecules and a data bank of small molecules to identify whether those small molecules bind well to their respective protein target. Small molecules that exhibit binding with sufficient affinity (otherwise known as a "hit") may be forwarded to secondary testing. Given the current level of computational power, including thousands of clustered PCs, millions of small molecules can be screened in just a few days. Ultimately, researchers will save both time and money by concentrating their efforts on a smaller number of bona fide drug candidates. An excellent example of a vHTS compound library is ZINC. (11).
3.2 ADMET Prediction
The ADMET Lab 2.0 was created using Django-Python and was then hosted on the Aliyun Elastic Compute Service running Ubuntu and using the Nginx web server as its web hosting. In order for the Django application to communicate with the Nginx web server, we used uwsgi as the interface between them. The application was built using the Model-View-Template (MVT) framework. The model layer represents the relationship between database entities and the business entities. The view layer contains the business logic to upload and download files, retrieve the correct information from the template layer, and execute access to the Deep Learning models. The template layer is where you will find many of the reports generated by ADMET Lab 2.0 including results visualization, some page rendering, documentation integration, etc. On the server reside two types of models. One type is a pre-trained model and another is an untrained model. The pre-trained models generate predictions using a deep learning solution. The programming language used to build the prediction generating models was Python. We used two different deep learning algorithms, PyTorch and DGL to create the generation algorithms. The RD Kit package also provided a variety of cheminformatics support. The server has been tested successfully on the latest version of Apple Safari, Google Chrome, and Mozilla Firefox. (1).
3.3 Molecular Modeling:
Molecular modelling is one of the main components of the CADD process and involves using computer technologies to represent how molecules behave. This is frequently accomplished through the construction of three-dimensional models of molecular structures comprising both protein and ligand molecules. This technology enables scientists to gain a clear insight into the structure and function of molecules, making it easier for them to ascertain how drugs may perform in the body. Through the provision of enhanced visualisation and analysis of how drug candidates interact with their respective target proteins, such as the determination of appropriate binding sites, scientists are aided in the design and development of novel pharmaceutical products. (12). Table 1 shows several new AI/ML-driven tools.
Table 1. Structures predicting methods and tools.
|
Methods |
Programs |
|
Homology Modeling: Utilize the established structure of homologous protein to construct the three-dimensional model of the target protein. |
MODELLER, SWISS-MODEL, Phyre2, RaptorX, I-TASSER |
|
Ab Initio Modeling: Construct a three- dimensional model of target protein by exploring its conformational space independently of experimental evidence. |
Rosetta, QUARK, AlphaFold, ESMFold, PCONS5 |
|
Threading: Align the protein sequence with the sequences of proteins whose structures are known to make a three dimensional model of the target protein. |
MUSTER, 3D-PSSM, LOMETS, HHpred |
|
Hybrid Modeling: Use two or more than two modeling methods together to make predicted structure more accurate. |
CABS-flex, PrimeX, GalaxyHomomer |
|
Molecular Dynamics: Use classical or quantum mechanics to model how the protein behaves over time. |
GROMACS, NAMD, CHARMM |
|
Knowledge based method: Use what you already know about protein structure and function to guess what the target protein's structure will be. |
ProSmoS, ProQ3D, I-TASSER-2GO |
|
Methods that don't use templates: Make 3D model of the target protein with not using templates or proteins that are similar to it. |
CONFOLD2, MetaPSICOV, TrRosetta |
|
Methods for fragment assembly: Put together pieces of known protein structures to make a 3D model of the target protein. |
PEP-FOLD3, Robetta, QUARK |
3.4 In silico ADMET (absorption, distribution, metabolism, excretion, toxicity) and drug safety prediction
The final selection and clinical development of new therapies expected to exhibit greater clinical efficacy will result from a careful and deliberate balancing of the drug-target interactions (potency and selectivity) and acceptable ADMET parameters. Data demonstrate that a drug molecule that enters Phase I clinical testing does so with less than a 10% chance of being marketed post completion of extensive preclinical development. Furthermore, significant long-term environmental impacts are being created due to the release of many pharmaceutical products during the later stages of drug development. The primary issues associated with the toxicity of pharmaceutical products are the human and environmental toxicity of pharmaceutical products (13). An orally active pharmaceutical product can be defined in terms of the following factors common to ADMET – i.e., the manner in which the drug is absorbed, distributed, metabolised, excreted, and ultimately considered to be toxic. (14). Lipinski's rule is associated with ADMET.
1) The maximum number of hydrogen-bond donors in a molecule is five (the sum of nitrogen-to-hydrogen bonds + oxygen-to-hydrogen bonds).
2) A molecule has ten hydrogen bond acceptors (all are either nitrogen or oxygen atoms).
3) The molecular weight (in grams) of a molecule must be less than 800 grams (or 500 daltons).
4) The octanol-water partition co-efficient (Log P) of a molecule must be less than 5.
5) The polar surface area (in Å2) of a molecule must be less than 190 Å2.
6) The number of atoms in a molecule can be between 20 – 70.
7) The maximum number of rotatable bonds in any one molecule is 10.
4. APPLICATION IN DISCOVERING NOVEL PLATFORMS
4.1 In silico autophagy methods and strategies in medicinal chemistry
Autophagy is a conserved cellular anabolic mechanism in all Eukaryotes, responsible for degrading and recycling proteins, organelles and damaged cellular material over extended periods. There is a now considerable body of experimental evidence from the past thirty years showing that autophagy has medical relevance as a potential target for medical investigation and as the basis of new therapeutic strategies in various disease models. In total, there are over 40,000 published studies on autophagy; however, there are a number of targets, mechanisms and processes of autophagy that have not yet been studied experimentally. The term "in silico autophagic methods" encompasses the large number of studies in the field of autophagy that employ web-based resources, mathematical models and systems-based methods as the primary means of studying autophagy. This editorial highlights newly developed methodologies and strategies for assessing autophagic processes by using in silico methods in medical research including, but not limited to, the identification of autophagic therapeutic targets and drug development. (15).
4.2 In-silico ADME Models
The ADME prediction process is complicated due to the many different physiological mechanisms that contribute to the properties we want to predict. This paper looks at how in-silico prediction of ADME processes can help direct medicinal chemists to make more favourable choices in the property space so that fewer compounds can be made to have the right biochemical and physicochemical properties. In addition to being able to predict the properties of a chemical compound using in-silico methods, these will also highlight physicochemical interdependencies between main physicochemical properties related to ADME in an effort to promote their optimisation. However, limitations of realism exist with these models when comparing them to in-vivo or in-vitro methods, therefore making them not a replacement for in-vivo or in-vitro studies. All of the in-silico models used in the world have been published in the literature and based on large diverse data sets. In this study, we review examples from each class of in-silico ADME models in order to show how useful these models can be when discovering new drugs. To illustrate the current state of the art for these ADME parameters, we will discuss the most current, relevant, and predictive examples available in the literature for every ADME category. Subsequently, we will provide a summary of each of the major model types developed for each class of in-silico ADME models, including rules, physicochemical properties, and 3-dimensional QSAR models and the overall predictive capabilities and SARs of these models. (16).
4.3 Drug Design Based on Structure
Many of the rational drug design projects at the company are based on computational approaches such as virtual screening and de novo design processes. NMR spectroscopy can provide molecular-level information about how ligands interact with their target molecules. This information is essential to understand how therapeutic agents interact with their targets and to develop new therapies. The group has studied how ligands bind to t-RNA and to the minor groove of DNA, including Hoechst 33,258, and will also be using NMR to study the interaction of ligands with proteins. The group has access to a variety of 500 MHz Bruker NMR spectrometers in the Department of Chemistry and has access to 300 MHz instruments in the School. (17).
5. BIOINFORMATICS IN CADD
A few years ago, the NIH (National Institutes of Health) established the BISTI (Biomedical Information Sciences and Technology Initiative), designed to help assess the current status of the United States' capacity to produce and advance bioinformatics. One area of research associated with CADD (Computer-aided Drug Design), which uses computer simulation to model how drugs interact with their receptor sites, has many overlaps with bioinformatics (14). As more protein targets become known due to advances in methods such as crystallography, NMR, and bioinformatics, there is a growing need for algorithms that can identify and analyze active sites on proteins, and predict new drug molecules that will selectively bind to these target sites. Creating new drugs can take a long time and cost a lot of money. On average, development of a new drug requires approximately 14 years of research and costs approximately $880 million prior to commercial availability. Therefore, it is necessary to use computers in each phase of drug development to accelerate or reduce the expense of developing new medications. (10).
6. RECENT ADVANCES
Pharmaceutical discovery and development's effectiveness hinges upon data accessibility. There is a great deal of biological sequence, chemical compound and other data from scientific literature and case studies. Some of the more prominent databases gathering and organizing this information across the entire biomedical field are outlined below. There are hundreds to thousands of biological databases mentioned each year. In addition, new and innovative ways are being developed that use computational techniques to create combinatorial libraries. Therefore, they represent the main focus of drug design through the use of computer-aided drug design(6). Papers presented during this conference will illustrate how optimization has been used in various sectors of engineering/industry. We called for papers from interested participants and received many interesting and responsive submissions; however, we cannot include the majority of them in the workshop. Due to space and time limitations, we can only feature a limited number of accepted papers from the COMS 2013 workshop at the ICCS 2013 conference, which provides an up-to-date overview of cutting edge applications of computational optimization, modeling and simulation. The applications include numerous different types of functions. Thaher et al. examined the maximum convex sum algorithm used to measure environmental variables, while Yang et al. worked with multi-objective methods developed from floral patterns to achieve an optimal result. Finally, Zelazny et al. utilized simulated methods of optimization. (5).
The studies cited above show that computational modeling and optimization can be used in many different ways. There will definitely be more applications in the near future. This workshop (5) gives people a chance to talk about and make progress in modeling and optimization.
7. CHALLENGES AND LIMITATIONS
While local models have a limited application relative to global models, when a classification model is changed to a different chemical series, it is likely that an estimated similarity will no longer hold. Conversely, global models will provide poor estimates for a test molecule that is very different than the group of test molecules used to create the model. These issues typically arise with QSAR models because QSAR are often considered to be local models. Therefore, the lead hopping mechanism discussed previously will only occur within new structural series.
Based on the numerous in silico pharmacology modelling examples described within this report, global models are likely to provide the most accurate results for the specific application or research. Several studies have provided comprehensive explanations of both the implementation and use-cases within QSAR frameworks of models and associated methods used to calculate the outcomes related to QSAR predictiveness. (18).
The use of integrated computational tools is changing how chemicals are discovered and how medicine is manufactured, and these types of tools have the potential to considerably speed up the creation of new drugs. However, there are many advantages and disadvantages to using these types of tools as well as integrating these types of tools, which must be carefully weighed in order for the methods to be effective when developing new drugs. Integrated methods are best suited for situations where biological and chemical data are both accurate and have great diversity. If either of the two are inaccurate or missing, predictions based on the two numbers may be less accurate than predictions based on accurate biological and chemical data together can produce. Therefore, scientists must have improved ways to collect and share both types of data so that they can accurately predict drug candidates based on that data. One major disadvantage of using integrative tools is that they are generally far more complicated than other types of computational tools. Furthermore, using integrative techniques to obtain information from multiple sources also makes it extremely difficult to perform calculations with the amount of data retrieved, consuming massive amounts of computer power; hence, as the number of data sources and data sets increases, so too does the amount of time needed to process the aggregated data. This process requires a relatively long period of time to develop new pharmaceuticals. In order to alleviate this issue with integrative research, algorithm and computer technology must continue to become more efficient and faster so that they can accurately process all of the complexities of integrative research. (4).
Because biological systems are so complex, this makes finding the right lead medication(s) a lot harder due to the fact that biological systems interact across three levels: Molecular, Cellular, and Organism. At each level, there is an intricate web of interactions. To find the right lead compounds, new integrative approaches will need to take into account the complexity of protein–protein interaction, how pathways communicate with one another, and how cells react to any number of things (as examples). Therefore, if we do not have complete knowledge about the underlying processes for all of these pathways then we are likely not going to be able to predict accurately. Understanding how biological systems function is necessary as it clearly guides us in determining how biological systems operate. Additionally, attempting to conceptualize “chemical space,” (which refers to the multitude of possible chemical compounds) is a daunting task. There are an infinite number of possibilities in terms of chemical compounds that can exist, which is difficult to comprehend. To fully take advantage of chemical space will require us to have new tools and methodologies in order to explore its vastness.
There are a number of unique options and characteristics that each of these new lead compounds can offer and therefore we will require future advances in computer programs in order to process through all of these options until we are able to find a lead compound that possesses the desired drug properties. In other words, we will require new levels of methodology in order to efficiently and effectively search through chemical space and find new lead compounds that will exhibit superior pharmacological properties. (4,19).
8. FUTURE VISION
Docking techniques and computer models for drug development are being used more than ever to discover novel medicines, allowing for improved activity of compounds and decreasing the likelihood of drugs failing at a later stage in their development process. These methods also enable designers to ascertain the location and manner of binding between the target protein receptor and the related ligand. Thus, this assists in creating more effective and specific analogues. Many people are now using these methods across multiple areas of drug discovery; including in silico target scanning, drug repurposing, polypharmacology, predicting adverse events and identifying potential "new chemical frameworks", in large compound libraries (20), and so on.
The principal objectives of QSAR and QSPR models are to predict the physical/chemical/biological properties of organic molecules through their molecular structures. These models allow scientists to better comprehend and design the pharmacological properties that are critical by concentrating on important chemical characteristics. QSAR models can be used to project how a drug will behave or interact throughout the early phases of drug development, thereby saving substantial time and money (not to mention effort); however, they cannot completely replace laboratory experiments. As computers become more powerful and diverse methodologies continue to be developed and additional data made available, QSAR modeling will become progressively more accurate and valuable. (5).
Some of the emerging computer technologies that are expected to have a significant impact on the future of drug development and discovering lead compounds include quantum computing, multi-omics data analyses, artificial intelligence (AI), and deep learning. As a result of advances in quantum computing, researchers may be able to understand how extremely complicated molecules interact in a much quicker timeframe. Similarly, deep learning algorithms could also provide researchers with much more accurate toxicity profiles of lead compounds and predictions on how lead compounds will interact with their target molecules (20).
Virtual screening will become a much more efficient and effective way to identify lead candidates. Another key area in which we will be better able to relate diseases to one another is through network pharmacology. In silico clinical trials will provide researchers with the opportunity to assess the likelihood of success, as well as to reduce their risks and costs, by providing them with data on how patients are likely to respond before conducting actual clinical studies. With the development of explainable AI, individuals will have an easier time interpreting predictive models, and collaborative efforts among various scientific groups will encourage governments to make more data available for research purposes and to collaborate on the development of new therapies. Finally, it will also be important for researchers to take steps to protect the confidentiality of their patients' data, and that the use of AI in a responsible manner is necessary to ensure proper use of AI (5).
To achieve this, researchers must continue to enhance their knowledge and skills in data science, artificial intelligence, and computational pharmacology to be able to better comprehend the complexities of drug development. (21).
CONCLUSION
The application of computational modeling, molecular docking, and bioinformatics has transformed the way pharmaceuticals can be researched and optimized in an extremely efficient way. They allow predicting how medications will interact with their targets and the way they are transported in the body. Moreover, these techniques are highly time-saving and cost-efficient compared to other means of conducting experiments. The use of molecular docking, QSAR, and QSPR methods provides a lot of knowledge concerning molecular behavior and structure. This, in turn, allows producing drugs which are not only effective but also harmless to patients' health(22).
There are some other new promising technologies such as quantum computing, machine learning, and artificial intelligence that can be used to accelerate and optimize the processes of pharmaceuticals' creation. Thanks to this set of methods, it is possible to find the new approaches to using medicines, evaluate their side effects, simulate their reactions on the organism, etc. Also, the use of network pharmacology along with multi-omics increases our understanding of diseases'(23).
In conclusion, it is possible to say that in-silico strategies prove to be rather useful for experimental research and will play an essential role in developing drugs during the first stages. With advances in computer (24).



Sourabh Khade, Dr. Shiv Shankar Hardenia, Dr. D. K. Jain, Computational Strategies in Drug Discovery: A Comprehensive Review of In Silico Approaches, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 1578-1592, https://doi.org/10.5281/zenodo.20567915
 10.5281/zenodo.20567915
10.5281/zenodo.20567915