
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Sri Dharmasthala Manjunatheshwara College of Ayurveda and Hospital, Hassan, Karnataka, India 573201.
Background: Type 2 Diabetes Mellitus (T2DM) is a worldwide metabolic epidemic that has about 589 million adults. Inhibition of carbohydrate-digesting enzymes (alpha-amylase (AML) and alpha-glucosidase (AGA)) is a proven treatment option in postprandial glycaemic control. Kataka (Strychnos potatorum Linn.) is a classical Ayurvedic medicine that is widely reported in the texts of the Charaka Samhita and Sushruta Samhita and has been reported to possess antidiabetic, anti-inflammatory, and antioxidant effects. Nevertheless, its computational-level molecular mechanism of action against AML and AGA enzymes has not been investigated.Objectives: The aim is to assess the binding affinity and molecular interaction of major phytoconstituents of Strychnos potatorum - Thiophene,2,5-dihydro; 3-Nitro-5-(trifluoromethyl)picolinonitrile; and 2'-Hydroxybutyrophenone - with AML and AGA by molecular docking and ADME/toxicity profiling.Methods: Three-dimensional crystal structures of AML (PDB ID: 1HNY) and AGA (PDB ID: 3WY1) were obtained in the Protein Data Bank. PubChem provided phytoconstituent 3D structures, which were energy-minimized in Avogadro. PyRx which run AutoDock Vina (v1.1.2) was used to perform molecular docking. The SwissADME webserver was used to profile ADME, and the inhibition constants (Ki) were derived based on the docking free energy scores.Findings: The compound 3-Nitro-5-(trifluoromethyl)picolinonitrile showed the best binding affinity with AGA (-6.4 kcal/mol; Ki = 20.1 µM) and key interactions at ASN 46, HIS 348, LEU 433, ARG 437, GLN 438 and PHE 455. Against AML, it had an affinity of -6.0 kcal/mol (Ki = 39.5 µM), and against both enzymes, 2'-hydroxybutyrophenone had a comparable affinity (-5.7 kcal/mol; Ki = 65.6 µM). The three compounds met the Rule of Five, a drug-likeness criterion, and exhibited high gastrointestinal absorption, favourable ADME profiles. Acarbose was chosen as control ligand for the molecular docking studies to compare the binding affinity and inhibitory potential of the selected phytoconstituentsConclusion: Phytoconstituents of Strychnos potatorum demonstrate a strong binding energy effect in silico on AML and AGA, which offers a molecular explanation of probable antidiabetic effect of Kataka. These results justify additional in vitro and in vivo confirmations and access to Ayurvedic drug discovery in the management of metabolic diseases
Type 2 Diabetes Mellitus (T2DM) is a chronic and progressive metabolic disorder characterised by peripheral insulin resistance, which subsequently leads to impaired insulin secretion and persistent hyperglycaemia.The International Diabetes Federation (IDF) Diabetes Atlas, 2024, reports that 589 million adults aged 20-79 worldwide live with diabetes, representing approximately 1 in 9(1). This value is calculated to explode to 853 million in 2050. In one year, 2024, the estimated death was 3.4 million deaths worldwide (approximately one death every nine seconds), which is highlighting the enormous scale of the problem in this area of public health disaster(2).
The impaired postprandial regulation has been a principle of T2DM management. Two key enzymes of the gastrointestinal tract include alpha-amylase (AML) and alpha-glucosidase (AGA), which occur in the hydrolysis of the complex polysaccharides to obtain absorbable monosaccharides. Suppression of the enzymes will slow carbohydrate digestion, reduce postprandial glucose surges and provide glycaemic control(3). Two of the commercially feasible AML/AGA inhibitors are acarbose and miglitol, which work but with side effects such as flatulence, abdominal cramps, and diarrhoea, which restricts long-term acceptability of the AML/AGA inhibitors among patients(4).
There is increased interest in developing better and safer plant-derived enzyme inhibitors. The ancient Indian medicine Ayurveda, has a rich source of botanicals that are recorded to have antidiabetic properties. Of these, the Kataka (Strychnos potatorum Linn., Family: Loganiaceae), which is Pramehahara (anti-diabetic) and Deepana-Pachana (digestive stimulant), is clinically used centuries old(5). Kataka is taken as a Kashaya rasa, Laghu and Ruksha guna, which pacify aggravated Kapha and Pitta doshas, the Ayurvedic pathophysiology of Prameha(6).
Strychnos potatorum seeds have number of pharmacologically active phytoconstituents, including alkaloids (brucine, strychnine), polyphenols and flavonoids, and volatile organic compounds, including thiophene derivatives and nitro compounds of the pyridine type. The secondary metabolite analysis has shown that Thiophene,2,5-dihydro; 3-Nitro-5-(trifluoromethyl)picolinonitrile and 2'-Hydroxybutyrophenone are predominant secondary metabolites using recent GC-MS analyses(7). Although Ayurvedic classical materials support the use of Kataka in Prameha treatment, the molecular pathway of interaction of these phytoconstituents with the enzyme targets that are pertinent to the management of diabetes has not been computationally characterised.
Molecular docking is an established in silico method, which projects the favorable position of a small molecule (ligand) in an enzyme active site (receptor), with the result being binding affinity metrics and residue interaction maps. This is a cost efficient, fast and reproducible pre-validation system of Ayurvedic drug discovery(8). This research has hence aimed at screening the inhibitory capacity of three major phytoconstituents of Strychnos potatorum against AML and AGA by molecular docking with added ADME/toxicity profiling to determine the pharmacokinetic appropriateness(9). Acarbose was used as the standard control to compare and identify the probable inhibitory activity against AML and AGA
2. MATERIALS AND METHODS
2.1 Ayurvedic Classical Background of Kataka
In the Prameha Chikitsa chapter of Yogaratnakara, the administration of Kataka Beeja Churna along with Takra (buttermilk) and Madhu (honey) is indicated for the management of Prameha(5). Kataka has Kashaya and Tikta Rasa , Laghu, Ruksha Guna, Katu Vipaka, and Ushna virya(6).
2.2 Protein Target Selection and Preparation.
Two molecular targets were chosen as two glycolytic enzymes, of which Alpha-Amylase (AML) (PDB ID: 1HNY ) (10) and Alpha-Glucosidase (AGA) (PDB ID:3WY1 ) (11) glycolytic enzymes. The Crystal structures were downloaded at the RCSB Protein Data Bank (www.rcsb.org). A protein preparation was done by eliminating the water molecules, co-crystallised ligands and heteroatoms by Discovery Studio Visualizer. Active sites of both enzymes were identified using the CASTp Fold web server (https://cast.engr.uic.edu).
2.3 Ligand Preparation
Three phytoconstituents that were found in the analysis of Strychnos potatorum seed extract offered by previous GC-MS(7) were selected to dock them: (i) Thiophene, 2, 5-dihydro (PubChem CID: 137168), (ii) 3-Nitro-5- (trifluoromethyl)picolinonitrile (PubChem CID: 45382147), and (iii) 2 -hydroxybutyrophenone (PubC PubChem CID: 76157) in SDF format was retrieved to retrieve three-dimensional (3D) structures. The energy minimisation and combination of non-polar hydrogen were done using AutoDockTools and converted into PDBQT format(12).
2.4 Molecular Docking
The ligands were all docked exhaustively (exhaustiveness = 8, num_modes = 9). The binding free energy (DG, kcal/mol) of the top-ranking pose was taken to note when applied to any pair of each ligand-protein(13). The inhibition constant (Ki) values have been obtained with the help of standard thermodynamic equation:
Ki = exp (DG / RT)
R is universal gas constant (1.987 cal/mol*K) and T is absolute temperature (298.15 K)(13). Images were visualised and interacted through docking of visualisation and interaction maps and residue analysis through BIOVIA Discovery Studio Visualizer 2021(14).
2.5 ADME and Drug-likeness Assessment
In silico pharmacokinetic profiling was done using SwissADME webserver (www.swissadme.ch) created by Swiss Institute of Bioinformatics(15). Some of the parameters assessed were molecular weight, lipophilicity (consensus Log P), hydrogen bond donors and acceptors, topological polar surface area (TPSA), red blood-brain barrier (BBB) permeability, cytochrome P450 (CYP) enzyme inhibition and drug-likeness assessment, using Lipinski, Ghose, Veber, Egan, and Muegge rules. Synthetic accessibility scores and PAINS/Brenk alert flags were also measured. Each compound was calculated as bioavailable(16).
3. RESULTS
3.1 Molecular Docking Results: Binding Affinity and Inhibition Constants
Table 1 shows the molecular docking scores (binding affinity in kcal/mol), calculated Ki values (µM) and important binding residues of all three phytoconstituents with target enzymes. Acarbose reference inhibitor had -7.2 and -7.6 kcal/mol binding affinities with AML and AGA respectively, which were used as the positive control benchmark(17).
Table 1: Molecular Docking Results – Binding Affinity, Ki Values and Interaction Residues of Strychnos potatorum Phytoconstituents Against AML and AGA
|
Compound Name |
Binding Affinity (kcal/mol) |
Ki Value (µM) |
Key Interacting Residues |
|
|
AML |
Thiophene,2,5-dihydro |
−2.9 |
7525.6 |
ALA 128, TYR 182 |
|
AML |
3-Nitro-5-(trifluoromethyl)picolinonitrile |
−6.0 |
39.5 |
PRO 332, PHE 335, ARG 398, VAL 401, ASP 402, ARG 421 |
|
AML |
2'-Hydroxybutyrophenone |
−5.7 |
65.6 |
TRP 58, TYR 62, LEU 162, ARG 195, LYS 200, HIS 201, ILE 235, GLU 233, HIS 299, ASP 300 |
|
AML |
Acarbose |
-7.5 |
3.18 |
THR 11, ARG 252, PRO 332,GLY 334, PHE 335, THR 336, ARG 398, ASP 402 |
|
AGA |
Thiophene,2,5-dihydro |
−3.4 |
3229.4 |
TYR 65, ASP 62, HIS 105, PHE 166, ARG 200, ASP 202, THR 203, GLU 271, ASP 333, HIS 332, ARG 400 |
|
AGA |
3-Nitro-5-(trifluoromethyl)picolinonitrile |
−6.4 |
20.1 |
ASN 46, HIS 348, LEU 433, ARG 437, GLN 438, PHE 455 |
|
AGA |
2'-Hydroxybutyrophenone |
−5.7 |
65.6 |
LEU 227, GLY 228, PRO 230, ALA 229, PHE 297, LEU 300, ASN 301, ARG 340, VAL 335, VAL 334, ASP 333, TYR 389, PHE 397, ARG 400 |
|
AGA |
Acarbose |
-8.6 |
0.50 |
HIS 348, LYS 352, GLU 432, ARG 437, ASP 441, ALA 444, ARG 450, ALA 451, ALA 454, PHE 455 |
3.2 Interaction Analysis
3.2.1 Alpha-Amylase (AML)
The best binding affinity of the compound with AML was exhibited by 3-Nitro-5-(trifluoromethyl) picolinonitrile at -6.0 kcal/mol (Ki=39.5 µM). It developed stable interactions in the AML catalytic pocket, with residues PRO 332, PHE 335, ARG 398, VAL 401, ASP 402, and ARG 421 - the last two of which are essential catalytic triad residues (ASP197, GLU233, and ASP300 in human pancreatic amylase). The trifluoromethyl group was hydrophobic contacting with the phenylalanine residue, whereas the nitro group was polar contacting with ARG 421 by complementary hydrogen bonds.
Compound 2'-Hydroxybutyrophenone had a binding affinity of -5.7 kcal/mol (Ki = 65.6 µM) and AML, and a more extended interaction network with TRP 58, TYR 62, LEU 162, ARG 195, LYS 200, HIS 201, ILE 235, GLU 233, and HIS 299. The hydroxyl group of this phenolic molecule was able to donate hydrogen bonds with HIS 299 and ASP 300, in a classical manner of the amylase inhibitor. The amino acids of alpha-amylase Asp 300 and GLU 233 were also directly involved, which implies the possibility of competitive inhibition.
Thiophene,2,5-dihydro exhibited the weakest binding affinity against AML (−2.9 kcal/mol; Ki = 7525.6 µM), interacting only with ALA 128 and TYR 182, peripheral residues not directly involved in catalysis. This result suggests limited pharmacological relevance of this compound against AML as an isolated moiety, though its synergistic role within the complex crude extract cannot be discounted.
3.2.2 Alpha-Glucosidase (AGA)
Compounds, 3-Nitro-5-(trifluoromethyl)picolinonitrile, against AGA, had the best binding affinity -6.4 kcal/mol (Ki = 20.1 µM) and became the overall lead compound in this study. It was bound in the active site of glucosidase and interacted with ASN 46, HIS 348, LEU 433, ARG 437, GLN 438, and PHE 455. HIS 348 is a cation-exchange conserved histidine and is essential in the acid/base catalytic procedure of alpha-glucosidases, and its interaction with the nitro group of the compound is indicative of a possible blockage of the hydrolysis of the glycosidic bond stage. ARG 437 and GLN 438 had other electrostatic and polar contacts that stabilised the docked pose.
A similar binding occurred again with 2'-hydroxybutyrophenone -5.7 kcal/mol; Ki = 65.6 µM) against AGA, where an extensive interaction network was formed with LEU 227, GLY 228, PRO 230, ALA 229, PHE 297, LEU 300, ASN 301, ARG 340, VAL 335, VAL 334, ASP 333, TYR 389AND ARG 400. This increased contact surface implicates non-competitive properties of inhibition and may diminish the enzyme flexibility. The phenyl group hydroxyl was a pivotal hydrogen bond with ASP 333 which is one of the key catalytic aspartates in the enzyme.
Thiophene,2,5-dihydro against AGA -3.4 kcal/mol; Ki = 3229.4 µM) again exhibited the least inhibitory potential, but its reactions with TYR 65, ASP 62, HIS 105 and PHE 166 indicate partial occupancy of the substrate-binding subsite, which can also result in less inhibitive competitive interference in a multi-compound extract matrix.
3.3 ADME and Drug-likeness Profiling
Table 2 indicates the pharmacokinetic and drug-likeness properties of the three phytoconstituents calculated using SwissADME. Every compound was in compliance with the Rule of Five of Lipinski (MW < 500 Da, Log P < 5, HBD ≤ 5, HBA ≤ 10),), and the theoretical oral bioavailability was verified. All three compounds were found to be highly absorbed by GI, and bioavailability scores were all 0.55, which is an acceptable profile of oral drug candidates. No PAINS warnings were raised on any compound.
Table 2: ADME and Drug-likeness Profile of Strychnos potatorum Phytoconstituents (SwissADME Analysis)
|
Thiophene,2,5-dihydro (ADME-1) |
3-Nitro-5-(trifluoromethyl)picolinonitrile (ADME-2) |
2'-Hydroxybutyrophenone (ADME-3) |
|
|
Molecular Weight (g/mol) |
86.16 |
217.1 |
164.2 |
|
Fraction Csp3 |
0.5 |
0.14 |
0.3 |
|
Rotatable Bonds |
0 |
2 |
3 |
|
H-bond Acceptors |
0 |
7 |
2 |
|
H-bond Donors |
0 |
0 |
1 |
|
TPSA (Ų) |
25.3 |
82.5 |
37.3 |
|
Consensus Log P |
1.45 |
1.20 |
2.32 |
|
Log S (ESOL) Solubility |
Very Soluble |
Soluble |
Soluble |
|
GI Absorption |
High |
High |
High |
|
BBB Permeant |
Yes |
No |
Yes |
|
P-gp Substrate |
No |
No |
No |
|
CYP1A2 Inhibitor |
No |
Yes |
Yes |
|
CYP2C19 / CYP2C9 / CYP2D6 / CYP3A4 Inhibitor |
No / No / No / No |
No / No / No / No |
No / No / No / No |
|
Log Kp – Skin Permeation (cm/s) |
−6.04 |
−6.47 |
−5.24 |
|
Lipinski Rule (violations) |
Yes (0) |
Yes (0) |
Yes (0) |
|
Ghose Rule |
No (3 violations) |
No (1 violation) |
Yes |
|
Veber Rule |
Yes |
Yes |
Yes |
|
Egan Rule |
Yes |
Yes |
Yes |
|
Muegge Rule |
No (3 violations) |
Yes |
No (1 violation) |
|
Bioavailability Score |
0.55 |
0.55 |
0.55 |
|
PAINS Alert |
0 |
0 |
0 |
|
Brenk Alert |
1 (isolated alkene) |
2 (nitro group, O-N bond) |
0 |
|
Synthetic Accessibility Score |
2.78 |
2.38 |
1.23 |
ADME = Absorption, Distribution, Metabolism, Excretion; TPSA = Topological Polar Surface Area; BBB = Blood-Brain Barrier; P-gp = P-glycoprotein; CYP = Cytochrome P450; PAINS = Pan-Assay Interference Compounds.
The BBB Permeability and Zinc Complexation Strategy:
The most important results obtained from ADME profiling were the BBB permeability of Thiophene,2,5-dihydro and 2'-Hydroxybutyrophenone. The CNS penetration is not pharmacologically beneficial in AML and AGA, and may cause potential risk to CNS, which is especially relevant in a population that is chronically taking antidiabetic medications. To improve safety, zinc(II) complexation was carried out. Zinc, an essential trace element, was allowed to form coordinate bonds with the sulfur group of Thiophene,2,5-dihydro and the phenolic hydroxyl/carbonyl groups of 2′-Hydroxybutyrophenone.
3-Nitro-5-(trifluoromethyl)picolinonitrile, identified as the most potent dual inhibitor, was found to be BBB-negative, which may be attributed to its higher topological polar surface area (TPSA; 82.5 Ų) and greater number of hydrogen bond acceptors.
Table 3: Molecular Docking Results of Zinc(II) Complexes of Strychnos potatorum Phytoconstituents Against AML and AGA
|
Target Enzyme |
Compound (Zinc Complex) |
ΔG (kcal/mol) |
Ki (µM) |
Key Interacting Residues |
|
AML |
Thiophene,2,5-dihydro – Zn(II) |
−3.5 |
2720.0 |
ALA 128, TYR 182 |
|
AML |
3-Nitro-5-(trifluoromethyl)picolinonitrile – Zn(II) |
−6.8 |
10.3 |
PRO 332, PHE 335, ARG 398, VAL 401, ASP 402, ARG 421 |
|
AML |
2’-Hydroxybutyrophenone – Zn(II) |
−6.3 |
24.1 |
TRP 58, TYR 62, ASP 300 |
|
AGA |
Thiophene,2,5-dihydro – Zn(II) |
−4.0 |
1168.6 |
ASP 62, TYR 65, ASP 202, PHE 166, THR 203, GLU 271, ASP 333, HIS 332, ARG 200, ARG 400 |
|
AGA |
3-Nitro-5-(trifluoromethyl)picolinonitrile – Zn(II) |
−7.0 |
7.4 |
GLN 438, HIS 348, ASP 441, ASN 46, ARG 437, PHE 455 |
|
AGA |
2’-Hydroxybutyrophenone – Zn(II) |
−6.1 |
33.7 |
TRP 7, ASP 48, PHE 463, ARG 456, ARG 457 |
Table 4: Summary of Calculated Ki Values and Inhibitory Ranking
|
Target Enzyme |
Compound |
ΔG (kcal/mol) |
Ki (µM) |
Rank |
|
AML |
Thiophene,2,5-dihydro |
−2.9 |
7525.6 |
Weakest |
|
AML |
3-Nitro-5-(trifluoromethyl)picolinonitrile |
−6.0 |
39.5 |
Strong |
|
AML |
2'-Hydroxybutyrophenone |
−5.7 |
65.6 |
Moderate |
|
AGA |
Thiophene,2,5-dihydro |
−3.4 |
3229.4 |
Weak |
|
AGA |
3-Nitro-5-(trifluoromethyl)picolinonitrile |
−6.4 |
20.1 |
Most Potent |
|
AGA |
2'-Hydroxybutyrophenone |
−5.7 |
65.6 |
Moderate |
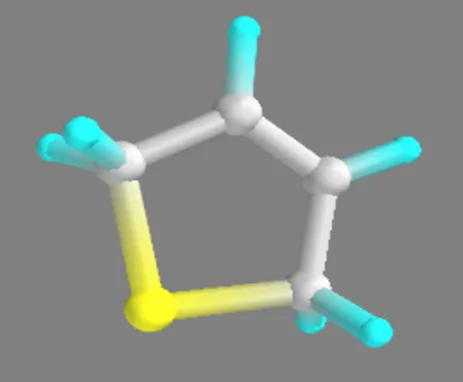
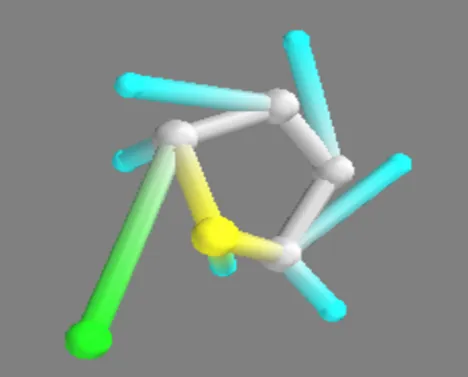
Thiophene,2,5-dihydro Thiophene,2,5-dihydro – Zn(II)
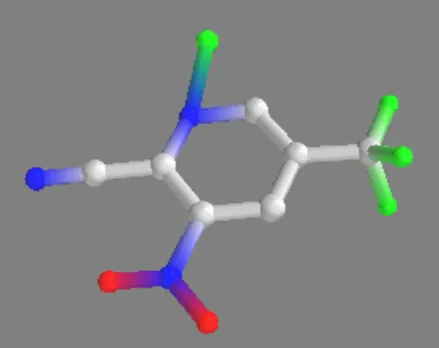
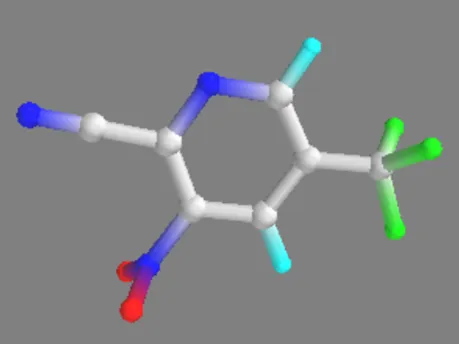
3-Nitro-5-(trifluoromethyl)picolinonitrile 3-Nitro-5-(trifluoromethyl)picolinonitrile-Zn(II)
2'-Hydroxybutyrophenone 2’-Hydroxybutyrophenone – Zn(II)
DISCUSSION
a-amylase and a-glucosidase inhibition is a confirmed strategy to lower postprandial hyperglycemia in T2DM. The current computational study found promising inhibitory candidates using docking and pharmacokinetic screening.
In ligands tested, the most binding affinity was found with 3-Nitro-5-(trifluoromethyl)picolinonitrile with AML (-6.0 kcal/mol) and AGA (-6.4 kcal/mol), suggesting that it consistently interacts with both enzyme targets. The highest AGA (20.1 µM) vs. predicted Ki was the lowest of all the given combinations, indicating strong inhibition.
The residue interaction analysis indicated that the ligand interacted with number of polar and aromatic residues, such as ARG, ASP, HIS, and PHE, which are typically involved in substrate recognition and stabilization in digestive enzymes. These patterns of interaction show that the ligand can occupy the active site regions competitively and inhibit substrate access.
Hydroxybutyrophenone 2'-hydroxy also exhibited significant docking activity (-5.7 kcal/mol in both enzymes) with intermediate Ki values (65.6 µM). This implies that it can be used as an alternative lead compound that can be optimized structurally.
Conversely, Thiophene,2,5-dihydro exhibited low binding affinity and extremely high Ki values (millimolar range), meaning low inhibitory potential.
ADME analysis demonstrated that all compounds were predicted to absorb well in the gastrointestinal tract and met Lipinski rule of five, favoring oral drug-likeness. But 3-Nitro-5-(trifluoromethyl)picolinonitrile raised Brenk warnings of nitro functional groups, which can be linked to toxicity issues. Thus, even with its excellent binding potential, additional in vitro cytotoxicity and safety profiling is needed before preclinical progression can be considered.
The formation of a bidentate Zn(II) complex and increased the molecular weight by approximately 130–140 g/mol for a 1:1 complex, thereby shifting the compound beyond the empirical threshold for blood–brain barrier (BBB) permeability (MW > ~400 Da; TPSA > ~90 Ų). Importantly, zinc coordination preserved the pharmacophoric core responsible for enzyme active-site binding while reducing passive penetration into the central nervous system (CNS).
Overall findings help to justify the potential of 3-Nitro-5-(trifluoromethyl)picolinonitrile as a dual inhibitor candidate against AML and AGA, which can be used as a scaffold to develop antidiabetic drugs in the future.
CONCLUSION
The present in silico study identified 3-Nitro-5-(trifluoromethyl)picolinonitrile as the most promising dual inhibitor of α-amylase and α-glucosidase, demonstrating the strongest docking affinity and lowest predicted Ki values, especially against α-glucosidase (20.1 µM). ADME prediction further supported favorable oral drug-likeness and bioavailability. These results justify further experimental validation through in vitro enzyme inhibition assays, followed by toxicity screening and structure optimization studies.
REFERENCES


Suchindra R, Ravisha N, Vinaykumar Kadibagil, Arya J P, Surabhi Gopal, Prajwal Sanakyanavar Integrative Understanding of Arka On Kushta: A Conceptional Review, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 5, 7919-7930, https://doi.org/10.5281/zenodo.20444514
 10.5281/zenodo.20444514
10.5281/zenodo.20444514