
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1Research Scholar, Guru Nanak College of Pharmaceutical Sciences, Dehradun
2Associate Professor, Guru Nanak College of Pharmaceutical Sciences, Dehradun
The translation of nanomedicines from laboratory research to clinical practice is critically dependent on the establishment of robust, globally aligned regulatory frameworks. This document provides a comprehensive analysis of the current regulatory landscape for nanotechnology-enabled health products (NHPs), with a primary focus on the frameworks established by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), and China’s Center for Drug Evaluation (CDE). The analysis begins by defining nanomedicines and exploring their transformative potential in diagnosis and therapy, including enhanced drug delivery, targeted therapeutic action, and improved bioavailability. It then identifies the fundamental mismatch between traditional regulatory paradigms and the unique properties of nanomaterials—such as increased surface area, altered reactivity, and complex biological interactions—which has resulted in critical gaps in manufacturing scalability, long-term safety assessment, and internationally standardized protocols. The core principles of a risk-based regulatory framework are examined in depth, emphasizing the FDA’s tiered approach to risk classification, the EMA’s hazard-focused paradigm, and the CDE’s emphasis on full-process quality control. A detailed breakdown of the FDA’s April 2022 guidance—“Drug Products, Including Biological Products, that Contain Nanomaterials”—is provided across its ten key subsections: description of nanomaterial(s), quality attributes and structural characterization, physicochemical characterization methods, dissolution/in vitro drug release methods, manufacturing process and in-process controls, excipients (function and safety), stability, postmarket CMC changes, nonclinical studies (ADME and route-specific risks), and clinical development. Additional relevant FDA guidances on liposomes and nanotechnology considerations are summarized, alongside the roles of the Center for Drug Evaluation and Research (CDER) and the Center for Biologics Evaluation and Research (CBER). The document further addresses the most pressing global challenges: the absence of harmonized definitions and classifications (contrasting the EU’s legally binding 2022 definition with the FDA’s property-driven “points to consider” and China’s structured classification system), profound methodological gaps in characterization and testing (including unreliable in-vitro-in-vivo correlation models), and the prevailing case-by-case assessment paradigm, which, while scientifically defensible, introduces unpredictability and delays. Finally, concrete recommendations for regulatory preparedness and global collaboration are presented, including the development of standardized reference materials and validated test methods, enhanced harmonization through the International Pharmaceutical Regulators Programme (IPRP) Nanomedicines Working Group, adoption of new approach methodologies (NAMs) for next-generation risk assessment, proactive early engagement with developers, integration of Quality by Design (QbD) principles, and implementation of advanced horizon scanning to anticipate emerging technologies.
The regulatory framework for nanomedicine is not merely a technical detail in the backdrop of scientific advancement; it is a critical determinant of whether these powerful technologies can safely and effectively reach patients[1]. This foundational section will first establish what we mean by nanomedicines, explore their transformative potential, dissect the unique challenges they pose to traditional regulatory paradigms, define the scope of our analysis, and finally, map the divergent definitions of nanomaterials that are at the heart of global regulatory discord. This analysis is grounded in a comparative review of the current international regulatory landscape, which reveals that while nanomedicines are at the forefront of healthcare innovation, their clinical application is often hindered by a complex and lagging regulatory navigation system[2][3]. The term "nanomedicine" is itself a broad umbrella, defined as the medical application of nanotechnology, or the use of nanoformulations to diagnose, prevent, and treat diseases. More specifically, from a regulatory perspective, the European Medicines Agency (EMA) typically considers a product to be a nanomedicine if it is a medicinal product—fulfilling the definition under European legislation—in which at least one component, be it the active substance or an excipient, is at a nano-scale size[4][5]. This convergence of nanotechnology with the health sciences has given rise to a new category of interventions often referred to as Nanotechnology-Enabled Health Products (NHPs)[6][7]. The foundational science behind this field is nanotechnology itself, an interdisciplinary domain that involves the design and application of materials with dimensions typically between 1 and 100 nanometers. The unique properties of these nanomaterials arise not just from their small size but from the resulting exponential increase in surface area relative to their volume; as bulk materials are broken down into nanoparticles, the cumulative surface area expands dramatically. This increased surface area enhances chemical reactivity, as a greater proportion of atoms are located on the surface, where they have fewer interatomic bonds and thus higher energy, leading to phenomena such as a significant decrease in melting point. These materials can be classified by their dimensions, including three-dimensional nanoparticles (like fullerenes), two-dimensional nanofibers and nanotubes, and one-dimensional structures like nanosheets and nanolayers, each offering distinct properties for biomedical applications[8][9]. In the regulatory domain, the sheer diversity of NHPs, from liposomal drug delivery systems to inorganic nanoparticles used in imaging, demands a nuanced, risk-based approach rather than a one-size-fits-all framework.
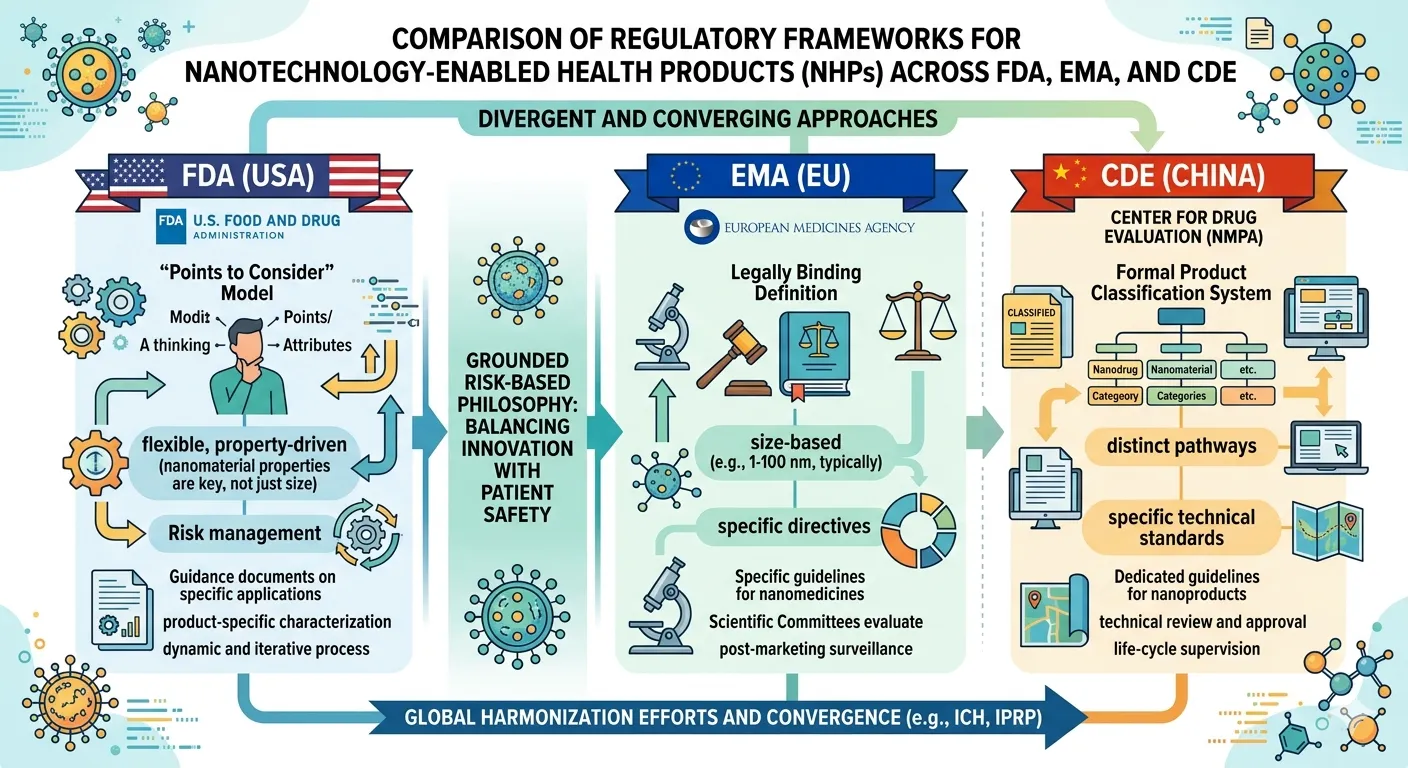
Fig: 1 Comparison of Regulatory Frameworks for Nanotechnology-Enabled Health Products (NHPs) across the FDA, EMA, and CDE.
The promises of nanomedicine in the realms of diagnosis and therapy are nothing short of revolutionary, and understanding this potential is crucial to appreciating the stakes of the associated regulatory challenges[10][11]. Autonomous nanomedicine represents a burgeoning branch of the field poised to revolutionize healthcare, moving beyond traditional methods by eliminating the need for external intervention to execute targeted actions within the human body. Traditional treatments like chemotherapy and radiotherapy are limited by non-specific toxicity and the development of drug resistance, issues that nanocarriers can overcome by enabling deeper drug penetration into thick tumor tissue while protecting healthy cells. These nano-sized carriers serve as specialized transport vehicles, navigating intricate physiological pathways to deliver therapeutic and diagnostic payloads with high precision[12][13]. By integrating automated nanoscale tools into detection processes, this technology offers the potential for swift and personalized health assessments, fundamentally reshaping disease management paradigms. The clinical potential spans a wide range of diseases, including cancer, cardiovascular conditions, and neurological disorders, aiming to improve patient outcomes while potentially reducing adverse reactions and healthcare costs. The economic and public health impact is also projected to be profound, shifting the focus from expensive late-stage disease management to more cost-effective, early-stage intervention. For instance, FDA-approved liposomes, which are small spheres of lipids around 100 nanometers in diameter, can encapsulate drugs, gene therapies, or oligonucleotides, offering a way to deliver therapies directly to their targets with high efficiency. However, while initial predictions of a "magic bullet" era in medicine have been moderated over the past two decades, the field has fulfilled some core promises, particularly in improving the delivery of poorly water-soluble drugs and enhancing the efficacy and biocompatibility of nano-based devices, demonstrating a tangible, albeit gradual, realization of its potential[14]. Despite the immense promise, the integration of nanomedicines into mainstream healthcare is impeded by a significant regulatory challenge: the frameworks governing them are lagging behind the astonishing pace of scientific advancement. The core issue is that the existing regulatory structures for pharmaceuticals, biologics, and medical devices were not originally designed to account for the complex, emergent properties of nanomaterials, creating a fundamental mismatch between scientific reality and regulatory expectations[15][16]. This mismatch is a primary driver of "critical gaps," which include unclear regulatory pathways, especially for advanced systems like nanorobotics, along with significant hurdles in manufacturing scalability and a lack of complete safety assessment protocols for long-term biocompatibility[17][18]. The regulatory landscape is further complicated by the "unavailability of an internationally standardized regulatory framework," leading to differences across global agencies that force developers to navigate a patchwork of often conflicting or inconsistent requirements. This situation not only creates barriers to entry and slows down the approval process but also introduces scientific uncertainty[19]. For example, there are currently no specific guidelines or standardized calculation methods for establishing an in-vitro-in-vivo correlation (IVIVC) for nanomedicines, a critical step in predicting performance and gaining approval, which represents a significant constraint in bringing these products to market. The nanotechnology field is evolving more quickly than the regulatory landscape can adapt, and as a result, the scientific community and regulatory bodies have been playing catch-up for the past two decades, despite substantial efforts to develop appropriate test criteria and analytical methodologies. The complexity of nanomaterials poses a particular challenge for regulators, as the same nanoscale properties that confer therapeutic benefits can also raise novel safety concerns, complicating the formation of necessary legislations and requiring a cautious, case-by-case approach that is both scientifically rigorous and time-consuming[20]. In light of the above, this framework has a clear and defined scope and set of objectives. Its primary purpose is to dissect the current regulatory landscape for Nanotechnology-Enabled Health Products (NHPs), with a specific focus on the frameworks established by the European Union and the United States, as they represent two of the largest and most influential markets for pharmaceutical innovation[21]. The goal is to not only identify the applicable requirements and guidelines for meeting regulatory expectations but also to highlight areas where these frameworks diverge and where they converge, thereby mapping the complex terrain developers must navigate[22][23]. A central objective is to provide a comparative analysis that serves as a strategic roadmap for understanding the risk-based approach advocated by key regulators like the FDA, which emphasizes the need to evaluate materials not just by size but by their engineered properties and potential impact on safety and efficacy. Furthermore, this framework aims to illuminate the collaborative global efforts currently underway, such as those by the International Pharmaceutical Regulators Programme (IPRP) Nanomedicines Working Group, which is actively working to facilitate regulatory convergence, harmonization, and information sharing among international agencies[24]. It also incorporates insights from major EU-funded projects like REFINE (Regulatory Science Framework for Nano(bio)material-based Medical Products and Devices), which is pioneering a framework specifically for the risk-benefit assessment of these products, developing and validating new analytical methods requested by regulators to address the most pressing challenges in the field. By covering these various aspects and the specific regulatory challenges related to quality, non-clinical studies, and safety, this framework serves as a foundational reference for stakeholders, including product developers, regulatory scientists, and policymakers, to understand the current state of nanomedicine regulation and the trajectory of its future evolution[24][25].
A fundamental and persistent hurdle in achieving a cohesive global regulatory strategy for nanomedicines is the absence of a single, universally accepted definition of what constitutes a "nanomaterial." The table below summarizes the definitions and approaches of the three major regulatory bodies covered in this framework: the European Union, the United States, and China. Each has adopted a distinct perspective, leading to significant variations in how products are classified and regulated[26].
Table 1: Comparative Overview of Key Nanomaterial/Nanomedicine Definitions
|
Jurisdiction / Body |
Definition / Guidance |
Regulatory Status |
|
European Union (EC/EU) |
"Nanomaterial" is a natural, incidental, or manufactured material consisting of solid particles, either on their own or as constituents, with most particles in the 1-100 nm range. |
Legally binding definition used across multiple sectors (chemicals, cosmetics, medical devices, etc.). |
|
United States (FDA) |
No regulatory definition. Uses "Points to Consider" based on size and properties: 1) Engineered to have at least one dimension in the 1–100 nm range. 2) Engineered to exhibit properties/phenomena (e.g., increased reactivity) attributable to its dimension(s), even up to 1,000 nm (1 micron). |
Non-binding guidance (Points to Consider). Decision on whether a product "involves the application of nanotechnology" is made on a case-by-case basis. |
|
China (NMPA/CDE) |
Nanomedicines are categorized based on structure: Drug Nanocrystals, Carrier-based Nanomedicines (e.g., liposomes, polymeric nanoparticles), and Other Classes (e.g., antibody-drug conjugates with nano-scale features). |
Defined in regulatory guidelines. Provides a formal classification system with associated technical guidance for submission. |
The European Union, through its Joint Research Centre (JRC) and updated 2022 Recommendation, has taken the most legally concrete and cross-sectoral approach. The new definition makes it easier to identify nanomaterials as materials consisting of solid, incidental, or man-made particles where most are in the 1-100 nanometer range, with the express goal of creating a single, uniform definition across all relevant EU legislation, from chemicals (REACH) to medical devices[27][28]. However, even this detailed definition is not without its critics; for instance, the French Agency for Food, Environmental and Occupational Health & Safety (ANSES) argues that the 2022 definition is too restrictive and could lead to a regression in the protection of public health by potentially excluding some materials from classification.

Fig: 2 Example on how FDA guidances on the same general subject relate to each other
In contrast, the U.S. Food and Drug Administration (FDA) has deliberately and consistently chosen not to establish a regulatory definition for the term "nanomaterial". Instead, its 2022 guidance for industry, “Drug Products, Including Biological Products, that Contain Nanomaterials,” provides a set of "Points to Consider" to help determine if a product involves the application of nanotechnology. This framework considers two primary points: first, whether a material is engineered to have at least one external dimension or an internal/surface structure in the nanoscale range (approximately 1 nm to 100 nm). However, crucially, the FDA recognizes that unique properties or phenomena attributable to size can emerge at larger dimensions. Therefore, the second point asks whether a material is engineered to exhibit such properties, even if its dimensions are up to 1,000 nanometers (1 micron). This flexible, property-driven approach is risk-based, aiming to focus regulatory attention on materials that, regardless of a specific size cutoff, could alter a product's safety or efficacy. This guidance applies to a diverse array of products, from those treating cancer to inflammation and infection, and emphasizes that while the same standards of safety, efficacy, and quality apply, nanotechnology's novelty means stakeholders must carefully think through any unknowns[29].
China, through its National Medical Products Administration (NMPA) and Center for Drug Evaluation (CDE), has established a formal classification system for nanomedicines that is codified in its technical guidelines. The CDE categorizes nanomedicines into three distinct types to provide a structured framework for review. These include "drug nanocrystals" (active pharmaceutical ingredients in nano-sized particulate form); "carrier-based nanomedicines" (where APIs are encapsulated within or attached to nanocarriers); and "other classes" which encompass complex structures like antibody-drug conjugates that exhibit nano-scale features. The CDE's overall strategy for quality control is based on a risk assessment approach, focusing on how the quality properties of the nanomedicine affect its safety and efficacy, a principle that aligns with the risk-based philosophies of the FDA and EMA. In August 2021, the CDE issued three key technical guidelines, covering quality control, non-clinical pharmacokinetics, and non-clinical safety evaluation, providing a comprehensive and cohesive set of rules for developers in the Chinese market[30]. The NMPA has also specifically addressed "nano-devices," providing classification rules for medical devices that contain nanomaterials and contact the human body.
The global need for harmonization in the face of these diverging definitions has never been more acute. The lack of an internationally standardized regulatory framework is a primary barrier, as the significant differences between global regulatory landscapes create inefficiencies, increase costs, and delay patient access to innovative treatments. In response, several crucial international initiatives have emerged to foster collaboration and convergence[31]. The most prominent is the International Pharmaceutical Regulators Programme (IPRP) Nanomedicines Working Group (NWG), which was established to share non-confidential information and promote regulatory harmonization. As of 2025, the IPRP NWG includes 16 member parties from regulatory authorities around the world, such as the FDA, EMA, Health Canada, Japan's PMDA, and China's NMPA, working collaboratively. This group has undertaken detailed mapping of regulatory terminology and has established specialized subgroups, such as a working group on lipid nanoparticles, to reach consensus on specific, complex product classes. The EU's Joint Research Centre (JRC) has also played a pivotal role by publishing technical reports that map the terminology used across different regulatory agencies, analyzing 31 frequently used terms to highlight similarities and differences as a foundation for harmonization[35][36]. Furthermore, the international community is actively pursuing a standardization roadmap, with events like the "International Standardisation Roadmap for Nanomedicine" workshop, which identified the development of standard test methods and reference materials as a key priority for global stakeholders. This collective effort acknowledges that a fragmented regulatory landscape is ultimately detrimental to the field, and that a move toward science-driven, global convergence is essential for nanomedicine to fulfill its considerable promise[37].
Core Principles of a Risk-Based Regulatory Framework
Foundational Principles for Nanomedicine Regulatory Assessment
The regulatory governance of nanomedicines rests upon a set of core principles that collectively aim to balance the immense therapeutic promise of nanoscale technologies with the imperative of patient safety[38][39]. These principles—centered on risk-based evaluation, rigorous characterization, comprehensive quality control, and the use of standardized methods—constitute the intellectual and procedural bedrock of modern nanomedicine regulation across all major jurisdictions, including the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), and China's Center for Drug Evaluation (CDE).
The Risk-Based Approach: Balancing Innovation and Safety
At the heart of nanomedicine regulation lies the foundational principle of a risk-based approach. The FDA has been the foremost proponent of this philosophy, advocating a framework that prioritizes the identification and management of risks specific to nanomaterials, rather than applying a uniform set of requirements to all such products[40][41]. The FDA's guidance, "Drug Products, Including Biological Products, that Contain Nanomaterials," released in April 2022 and effective from late 2023, serves as the cornerstone of this approach. It recommends a risk-based evaluation of potential changes in exposure, safety, and effectiveness, encouraging manufacturers to categorize products into low, medium, and high-risk tiers based on nanomaterial characteristics and their intended routes of administration. This tiered strategy ensures that regulatory scrutiny is proportional to the potential hazard, allowing lower-risk products to proceed with less burdensome characterization while focusing intensive resources on those with higher uncertainty. The FDA's approach is not a separate regulatory pathway for nanomedicines but an adaptation of existing frameworks for drugs, biologics, and devices, applying heightened scrutiny to nanoscale properties within these established structures. The EMA adopts a complementary but distinct risk-prevention posture, emphasizing hazard identification, hazard characterization, exposure assessment, and risk characterization as its four-part risk assessment paradigm. Meanwhile, the European Commission's Joint Research Centre (JRC) has advanced a formal definition of nanomaterials to create uniform application across all relevant EU legislation, from chemicals to medical devices, reflecting a legally concrete approach to risk governance. China's CDE similarly advocates a risk assessment approach, but with a particular focus on how quality properties of nanomedicines affect safety and efficacy, emphasizing full-process quality control from raw materials to final product. This multi-jurisdictional alignment on risk-based assessment underscores the global consensus that nanomedicines require evaluation strategies proportionate to their unique characteristics.
Key Considerations for Nanomaterial Characterization
Effective regulatory oversight begins with comprehensive characterization, yet this remains one of the most significant challenges in the field. The FDA guidance outlines several key risk factors for assessment, including the adequacy of nanomaterial characterization, the complexity of nanomaterial structure, mechanistic understanding of physicochemical properties and biological effects, predictability of in vivo release, physical and chemical stability, maturity of manufacturing and analytical methods, and the route of administration. For any drug product containing nanomaterials, an exhaustive description of the nanomaterial(s) is paramount, encompassing essential characteristics such as size, charge, morphology, composition, and complexation, tailored to the product's life cycle stage[42][43].
Regional Regulatory Frameworks
United States FDA
The regulatory framework established by the U.S. Food and Drug Administration (FDA) for nanotechnology-enabled medical products is anchored in a final guidance for industry entitled “Drug Products, Including Biological Products, that Contain Nanomaterials,” issued in April 2022. This foundational document supersedes the 2017 draft version and reflects the agency’s current thinking on how existing drug and biologic regulations should be applied to products that incorporate engineered nanomaterials in their finished dosage form[45]. Central to the FDA’s approach is a risk-based framework designed to balance therapeutic innovation with patient safety. The guidance explicitly refrains from classifying nanomaterials as inherently benign or harmful. Instead, it focuses on identifying and managing potential risks introduced by nanoscale properties, while reaffirming that all nanomedicine products must meet the same rigorous standards of safety, efficacy, and quality as conventional drug products. A critical feature of the final guidance is the addition of a glossary of key terminology and updated sections for abbreviated new drug applications (ANDAs) and over-the-counter (OTC) monograph drugs, providing clearer pathways for generic and non-prescription nanomedicines.
Detailed Content Framework of the 2022 FDA Guidance
The 2022 guidance provides a comprehensive roadmap for product developers, mapping out detailed recommendations across the entire product lifecycle—from early characterization through post-market changes. Each of the ten subsections listed below addresses a critical aspect of nanomedicine development:
Description of the Nanomaterial(s)
Applications must include a thorough description of nanomaterials within the finished dosage form. This entails detailing physical characteristics such as size, charge, shape, morphology, composition, and complexation. The level of description should be appropriate to the product’s development stage and become more refined as additional data become available. Comprehensive description is the essential first step to enable the precise control over nanomaterial attributes needed for consistent product quality[46][47].
Nanomaterial Quality Attributes and Structural Characterization
The identification of critical quality attributes (CQAs) is paramount. Manufacturers must determine which physical and chemical attributes are essential to product quality and link them directly to function and potential impact on product performance. This requirement often extends to the nanomaterial’s internal architecture, including core-shell structures, surface modifications, and the spatial arrangement of multiple molecular components—all of which can profoundly influence biological interactions. Nanomaterial Physicochemical Characterization Methods The guidance stresses the critical need to justify all analytical methods used to measure nanomaterial properties, including size, surface charge (zeta potential), surface area, and purity. Developers must account for factors that can distort nano-scale measurements, including sample preparation artifacts and unintended interactions between the sample and analytical equipment. This is especially challenging given that standard light microscopes cannot resolve objects smaller than 250 nm, necessitating specialized techniques such as electron microscopy, dynamic light scattering (DLS), and nanoparticle tracking analysis (NTA).
Dissolution/In Vitro Drug Release Methods
This section clarifies how manufacturers should use dissolution or in vitro release testing (IVRT) methods for quality assessment to demonstrate how the manufacturing process and formulation influence the release of the drug product, which directly contributes to its clinical performance. The FDA acknowledges that standard compendial methods may be insufficient for nanomedicines and is open to consultations to develop product-specific, clinically relevant release methods.
Manufacturing Process and In-Process Controls
A key emphasis of the guidance is the need for robust chemistry, manufacturing, and controls (CMC) procedures. All drug products containing nanomaterials must be manufactured in accordance with current good manufacturing practice (CGMP). The guidance further recommends that manufacturers identify process controls early in development that directly relate to the CQAs of the drug product. As one analysis notes, “In 2022, the FDA Guidance on Drug Products containing Nanomaterials was published, and it provides a roadmap for submission of a nanomedicine drug product,” specifically highlighting the need for additional guidance on CMC issues for complex lipid-containing nanoparticles[48].
Excipients (Function & Safety)
The guidance explicitly addresses nanomaterials that serve as excipients (inactive ingredients). The safety and functional properties of these nano-excipients must be determined based on their intended purpose. For example, in a liposomal formulation, the purity of constituent lipids or the molecular weight of polymers in a drug delivery system can be critical attributes that must be controlled. Standard FDA guidance for evaluating excipient safety applies when a common excipient is deliberately modified into a nanomaterial.
Global Challenges, Gaps, and the Path Forward
The path of nanomedicine from the laboratory to the patient is obstructed by deep-seated regulatory challenges that form a persistent "translational gap". The absence of globally harmonized definitions and classifications stands as the primary barrier, creating a fragmented international landscape where each jurisdiction operates on its own terms. While a global consensus largely recognizes nanomaterials as those with dimensions between 1 and 100 nanometers, the EU legally enforces a binding definition anchored by a threshold where "50% or more of the constituent particles have at least one dimension in the size range of 1–100 nm", mandating a minimum of one nanoscale dimension, which is the most widely employed definition internationally. In contrast, the FDA deliberately avoids a statutory definition, instead requiring a risk-based assessment on a product-by-product basis. The introduction of the EU definition in 2022, which shifts the criterion from a "major fraction" of particles to "50% or more", further deepens the divide with the FDA's approach. Simultaneously, inconsistent classifications between medicinal products and medical devices force developers to navigate complex, multi-track regulatory pathways. This lack of a common regulatory language directly obstructs global pharmaceutical innovation by forcing manufacturers to tailor their data packages to multiple, incompatible jurisdictional requirements[48]. This definitional ambiguity is compounded by profound methodological gaps in nanomaterial characterization and testing. The nanomaterials employed in healthcare exist in countless "nanoforms," each varying in size, shape, surface chemistry, and solubility. Traditional toxicology methods are often insufficient for assessing nanomaterials; their unique behaviors—such as long-term accumulation in the liver and spleen, which can trigger chronic inflammation and provoke a reactive oxygen species (ROS) burst—remain dangerously under-characterized by conventional toxicity assays that are not designed to detect these delayed, accumulative effects. The literature is rife with discordant and conflicting results for nanomaterials, largely because a standardized and universally accepted "list of physicochemical parameters" to guide characterization does not exist. The inability to accurately predict in-vivo behavior from in-vitro data—specifically, the development of reliable in-vitro-in-vivo correlation (IVIVC) models for nanoformulations—remains a critical blind spot in the safety assessment of nanomedicines[50]. Consequently, the global regulatory paradigm for nanomedicine is not one of standardized rules but of a "case-by-case" assessment. While this approach is scientifically defensible—acknowledging that slight variations in nanoparticle size or surface chemistry can radically alter both therapeutic efficacy and safety—it has unintentionally introduced unpredictability and administrative delays for manufacturers. The lack of a formal, distinct regulatory classification for nanomedicines in many jurisdictions (including the United States) forces each product to be evaluated under a traditional framework that is ill-suited to the material's unique complexity. This paradigm is inherently reactive rather than proactive, chasing scientific innovation instead of anticipating it, and contributing to the "valley of death" that prevents many promising technologies from ever reaching the clinic. Even the Doxil® case, a landmark in nanomedicine, highlights the immense difficulty of this approach, as demonstrating "sameness" for generic nanomedicines proved to be a regulatory and scientific hurdle of the highest order. To overcome these obstacles and build a functional future, a series of concrete recommendations for regulatory preparedness and global collaboration must be implemented immediately. The highest priority is the development of standardized reference materials and validated test methods to anchor regulatory science, a conclusion reached unanimously at the 2023 "International Standardisation Roadmap for Nanomedicine" workshop hosted by the French National Metrology Institute (LNE) and supported by global stakeholders. There is an urgent need for harmonization between disparate agencies, with the International Pharmaceutical Regulators Programme (IPRP) Nanomedicines Working Group—which as of 2025 includes sixteen global regulatory authorities—serving as the most viable platform for this international coordination. It is essential for regulators to adopt new approach methodologies (NAMs) for next-generation risk assessment (NGRA), including in-silico models and high-throughput in-vitro assays, moving beyond obsolete animal studies. National agencies must proactively facilitate early engagement with developers; the UK's MHRA Innovation Office and the European Medicines Agency's "Horizon Scanning" initiative are effective models for structuring early-stage guidance and de-risking the development process before formal submission. Furthermore, academic researchers, manufacturing industries, and clinical gatekeepers must participate in ongoing standardization efforts, such as the new Working Group under CEN/TC 352, to ensure that technical requirements are feasible and grounded in real-world science. In parallel, the incorporation of Quality by Design (QbD) principles from the earliest stages of research would systematically identify critical quality attributes (CQAs) and process parameters, shifting the field from reactive troubleshooting to the proactive design of safe and effective products. Finally, regulatory science itself must be modernized through the implementation of advanced horizon scanning to preemptively identify emerging trends, allowing guidelines to be developed proactively rather than in response to market submissions. By systematically implementing these collaborative strategies, the global community can begin to solve the definitional, methodological, and structural challenges that have long hindered clinical progress.
CONCLUSION
The regulatory governance of nanomedicines stands at a critical inflection point. The therapeutic promise of nanotechnology-enabled health products—from targeted cancer therapies and advanced drug delivery systems to precision diagnostics—is undeniable. Yet, the realization of this promise is contingent upon the development of regulatory frameworks that are as sophisticated and dynamic as the technologies they seek to oversee. This analysis has demonstrated that while significant progress has been made, particularly with the FDA’s 2022 final guidance and the EMA’s horizon scanning initiatives, the global regulatory landscape remains fragmented, inconsistent, and, in key respects, scientifically lagging.
REFERENCES

Kashish Papneja*, Archana Rautela, Nanomedicine regulatory framework, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 5, 7080-7093. https://doi.org/10.5281/zenodo.20404064
 10.5281/zenodo.20404064
10.5281/zenodo.20404064