
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Shree Gulabrao Deokar College of Pharmacy ,Jalgaon,Maharashtra
Background: Pharmacovigilance as described by the World Health organizations as "the science and activities pertaining to the identification, evaluation, comprehension, and avoidance of negative consequences or any other issue pertaining to drugs a crucial part in guaranteeing that patients receive safe medications. Our It is possible to learn more about a drug's side effects. through a variety of methods, such as impromptu reporting, intentional database research and sive monitoring. New procedures, both are being developed at the scientific and regulatory levels in an effort to improve pharmacovigilance. On a These include risk and conditional approval at the regulatory level.management strategies; from a scientific perspective, openness and Two key components are increased patient involvement. Goal : To examine and talk about different facets of pharmacovigilance, including the development of new methods
A crucial area of medicine is pharmacovigilance, which focuses on identifying, evaluating, comprehending, and preventing side effects or other drug-related issues. Making sure medications are safe has become a top public health concern due to the growing use of medications across the globe. Drug safety monitoring is crucial because adverse drug reactions (ADRs) are a major cause of morbidity and mortality. Following past drug tragedies like the Thalidomide tragedy, which brought attention to the necessity of stringent drug monitoring systems, the idea of pharmacovigilance gained significance. Since then, international pharmacovigilance programs have been established by organizations such as the World Health Organization to enhance patient safety. Activities related to pharmacovigilance include gathering, identifying, evaluating, tracking, and preventing side effects related to pharmaceuticals. From clinical trials to post-marketing surveillance, it is essential to a drug's lifecycle. The Pharmacovigilance Programme of India, which attempts to protect public health by guaranteeing the safe use of medications, is responsible for coordinating pharmacovigilance in India. Physicians, pharmacists, and nurses are among the healthcare professionals who are crucial in reporting adverse drug reactions (ADRs) and enhancing the efficacy of pharmacovigilance systems. In order to improve signal detection and data analysis, pharmacovigilance is incorporating new techniques like artificial intelligence as a result of technological advancements. Despite these developments, problems like low awareness and underreporting of ADRs continue to be major obstacles. Thus, maintaining medication safety, enhancing therapeutic results, and safeguarding public health all depend on pharmacovigilance. Pharmacovigilance has changed in recent years due to technological developments. The use of technologies like artificial intelligence and big data analytics to swiftly and precisely identify safety signals is growing. The significance of real-time pharmacovigilance systems was brought to light by the monitoring of vaccines, particularly during the COVID-19 pandemic.
What is Already Known –
A well-known scientific field, pharmacovigilance is concerned with the identification, evaluation, comprehension, and avoidance of adverse drug reactions (ADRs). Drug safety monitoring has been reinforced over time by international systems like the WHO's Programme for International Drug Monitoring.
What This Study Adds –
This review paper builds upon existing knowledge and provides a comprehensive and updated perspective on pharmacovigilance by integrating traditional concepts with modern advancements.
LITERATURE REVIEW
|
Sr.No. |
Author & Year |
Research Focus/ Goal |
Key Finding |
Referance |
|
1 |
Rocca et al., 2019 |
Development and evolution of pharmacovigilance |
Pharmacovigilance evolved as a discipline combining pharmacology and epidemiology for drug safety assessment |
Rocca et al., 2019 |
|
2 |
Domagk, 1935 |
Effectiveness of sulfonamides in treating infections |
Sulfonamides became the first successful antibacterial drugs and raised the need for drug regulation. |
Morales and Bosch, 2007 |
|
3 |
Florence, 1960 |
Investigation of thalidomide toxicity |
Long-term thalidomide use caused paresthesia, muscle cramps, and neurological toxicity. |
Florence, 1960 |
|
4 |
Lenz, 1962 |
Relationship between thalidomide and congenital deformities |
Reported congenital abnormalities in children exposed to thalidomide during pregnancy. |
Lenz, 1962 |
|
5 |
Pfeiffer & Kosenow, 1962 |
Statistical association of thalidomide with birth defects |
Strong link established between first-trimester thalidomide use and fetal malformations. |
Pfeiffer and Kosenow, 1962 |
|
6 |
McBride, 1961 |
Detection of congenital abnormalities due to thalidomide |
First spontaneous report linking thalidomide to birth defects. |
First spontaneous report linking thalidomide to birth defects. |
|
7 |
WHO Programme for International Drug Monitoring, 1972 |
Global ADR monitoring system |
Established international pharmacovigilance collaboration for drug safety monitoring. |
WHO Technical Report, 1972 |
|
8 |
|
Use of linked healthcare databases for drug safety studies |
Use of linked healthcare databases for drug safety studies |
|
|
9 |
BNT162b2 COVID-19 Vaccine Studies, 2020–2022 |
Safety and reactogenicity of COVID-19 vaccines |
Clinical trials and post-marketing surveillance confirmed vaccine safety and effectiveness. |
BNT162b2 Vaccine Safety Review |
|
10 |
Pharmacovigilance Programme of India (PvPI), 2010 |
Pharmacovigilance Programme of India (PvPI), 2010 |
Established ADR Monitoring Centres across India for better drug safety reporting. |
PvPI Framework |
|
11 |
AI in Pharmacovigilance Research, Recent Years |
Use of artificial intelligence in ADR monitoring |
AI improved case processing, signal detection, and analysis of large safety databases. |
|
|
12 |
Veterinary Pharmacovigilance Studies, Recent Years |
Misuse and abuse of veterinary medicines |
Veterinary drugs such as xylazine and pentobarbital showed increasing abuse and fatality reports. |
|
HISTORY
The disciplinary field of pharmacovigilance (PV) is frequently associated with pharmacological and epidemiological research. This is due to the fact that it is primarily regarded as a discipline that focuses on evaluations in the area of medication safety and approval (Rocca et al., 2019). Nonetheless, Rocca et al. (2019) acknowledge that this field has produced several novel insights concerning epidemiology and epistemology. However, the development of new approaches, procedures, and evidence standards to facilitate the application of risk assessment is becoming more and more important.
The initial systems for pharmaceutical control: The sulfonamide situation in 1932, Domagk (1895–1964) proved that sulfonamides were effective in treating streptococci. Following the establishment of the trademark patent Protonsil in 1935, which made it possible to market the first medication containing this active ingredient, the first sulfonamide was produced and sold (Morales and Bosch, 2007). The first sulfonamides' effectiveness was widely reported in the media, which had a significant social impact. In actuality, the favorable perceptions of the drug were conditioned by an imaginary factor. The New York Times reported in 1936 that President Roosevelt's son had been treated with Prontylin, a dispensing form of Protonsil, following his hospital admission due to a severe tonsil infection caused by Streptococcus (Morales and Bosch, 2007). The Food and Drug Administration (FDA) became aware of the increasing regulatory issue caused by the growth of sulfonamides due to the commercial success of this medication (Cooper, 2002).
Pharmaceutical control system maturity: Thalidomide as an example Alpha-phthalimido-glutarimide, or thalidomide, was successfully obtained in 1954 by the German company Chemie Grünenthal. This medication was used in 1957 to treat anxiety, insomnia, nausea, and vomiting in pregnant women. It was categorized as a sedative and hypnotic (Martínez-Frías, 2012). Following thalidomide exposure, the first isolated case of phocomelia was reported in 1956. Over the next five years, 3,000 cases of dysmelias, congenital malformations like amelias, phocomelia, or absence/hypoplasia of the thumb or fingers, among other conditions, were progressively reported globally (Papaseit et al., 2013). However, Florence (1960) reports in a brief letter to the British Medical Journal that patients receiving thalidomide for long periods of time (8 months to 2 years) complained of the following side effects: (1) Paresthesia affecting first the feet and then the hands. (2) The extremities are cold, and the toes are noticeably paler. (3) Slight ataxia on occasion. (4) Leg muscle cramps during the night.
The adverse effects decreased when the patients stopped taking the drug and the treatment was discontinued. Leslie Florence became suspicious of thalidomide's toxicity as a result. A series of letters about the effects of thalidomide were subsequently published in The Lancet magazine in January 1962. Lenz (1962) signed the first of these letters, which described 52 children who had deformities as a result of their mothers consuming this substance while they were pregnant. However, Lenz notes in this letter that the author attended a conference on November 18, 1961, where the topic of this substance's role in the development of human malformations was previously discussed. Another letter by Pfeiffer and Kosenow (1962) was published in the same issue of The Lancet. In it, he stated that there was a strong statistical correlation between the use of thalidomide during the first trimester of pregnancy and the development of defects. Thalidomide is emphasized in the third letter, which is signed by Hayman (1962), the managing director of the Distillers Company, and starts by thanking them for the expressions of gratitude they received. He continues by saying that it is especially challenging to determine the negative effects of this substance because of the scant data and official statistics. The objective data from the different researchers demonstrated that thalidomide was not as safe as it was claimed to be, regardless of one's personal opinion of Hayman's writing.
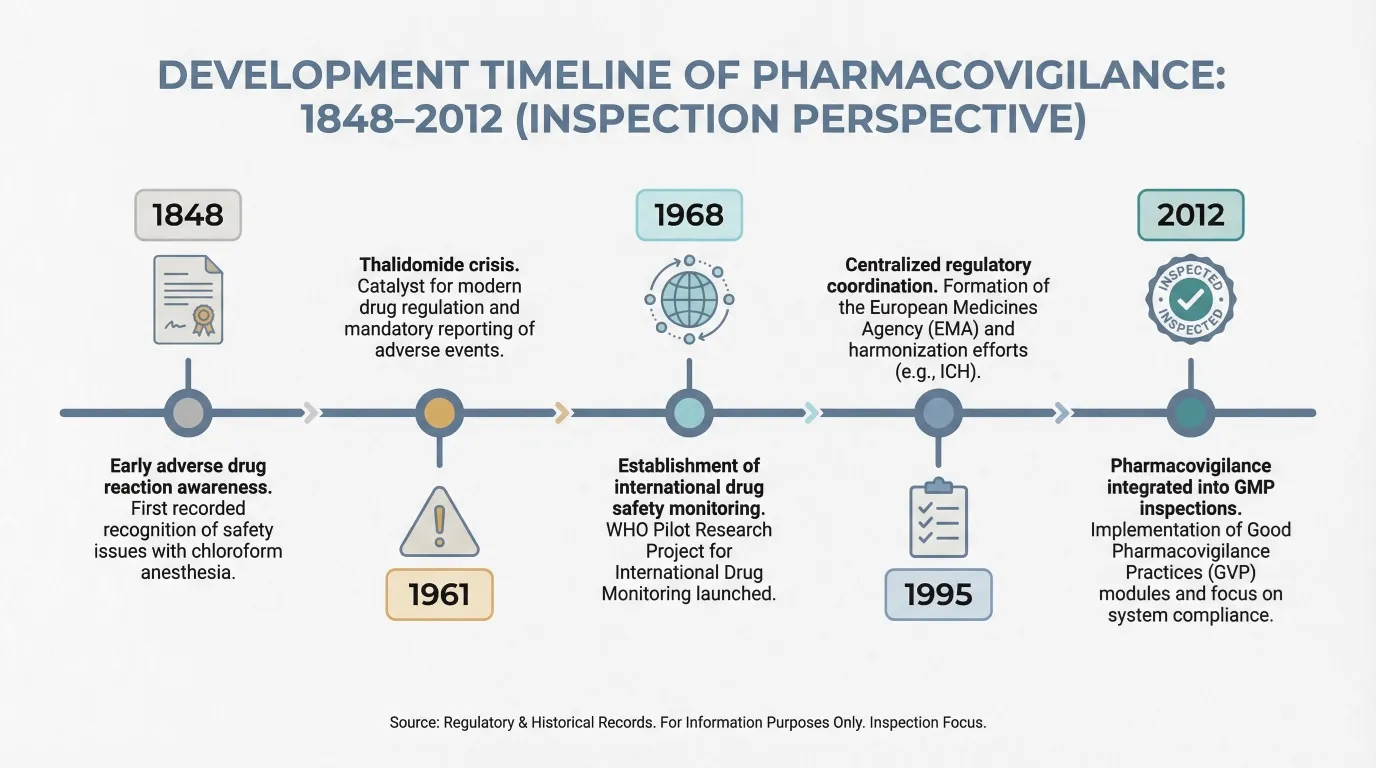
Fig No 1 : Development Timeline of Pharmacovigilance
METHODS USED IN PHARMACOVIGILANCE -
The actions carried out in the name of pharmacovigilance can be broadly separated into three categories: regulatory, both academia and industry. Pharmacovigilance that is regulated is motivated by the goal of giving medications a beneficial effect harm profile to the general public. Several issues pertaining to the topic of regulatory post-marketing surveillance will be covered in this background, then an explanation of the techniques employed to identify novel ADRs and an analysis of the benefits and drawbacks of each approach
Inadequate clinical trial data to assess medication risk Currently, the primary technique for obtaining data on a medication in the pre-marketing stage is to carry out a clinical trial. There are three types of pre-marketing clinical trials stages. Phase III research frequently uses double blind randomization controlled trials, which are thought to be the most methodical approach to establishing whether a cause-and-effect. There is a connection between an outcome and a treatment.But when it comes to keeping an eye on a medication's safety, The design of this study is not ideal. Because of the small number of patients taking part, it is typically not feasible to determine which ADRs are infrequent. The comparatively brief it is challenging to identify ADRs due to the length of clinical trials with an extended latency. Clinical trials also have the drawback of the population used to test drugs. The traits of the participants don't always match the features of the population that it will eventually be utilized; therefore, it could be challenging to extrapolate the findings from clinical trials to the general public at large. This is particularly true for women and the elderly or for members of a minority ethnic group. To investigate uncommon ADRs, long-latency ADRs, and ADRs in particular populations, it is crucial to closely monitor the medication during the post-marketing phase.
There are two types of descriptive research: Intense monitoring and impromptu reporting—will be talked about here. Analytical research can be carried out with a range of methods, such as case-control studies, clinical trials and cohort studies. To be able tocarry out case-control and retrospective cohort studies, data which have been routinely and reliably gathered must be accessible. To give an illustration of such studies, we outline two databases from Europe here. Frequently employed in analytical research, the General Practitioners UK Research Database (GPRD) and the PHARMO The Netherlands' Record Linkage System.Unplanned reporting. A 1961 letter from the Australian doctor WG The Lancet published McBride. In this correspondence, he revealed his finding that infants whose mothers had utilized thalidomide during pregnancy were born with congenital anomalies more frequently than infants who had not been thalidomide exposure during pregnancy. In the years ahead, it became apparent that thousands of newborns had been born with limb deformities brought on by the mother's use of thalidomide. To stop a similar catastrophe from systems were established worldwide with the goal of controlling and keeping an eye on drug safety. Systems for spontaneous reporting (SRS) were developed, and these are now the main way that post marketing data regarding medication safety. The primary SRS's role is to identify new signals early on. uncommon and dangerous ADRs. A system of spontaneous reporting permits doctors and, more frequently, pharmacists and patients to notify a pharmacovigilator of any suspected adverse drug reactions. lance center. The pharmacovigilance task center is to gather, evaluate, and disseminate the reports stakeholders of the possible danger when indications of new ADRs occur.
In the past, signal detection in spontaneous reporting has mainly occurred on the basis of case-by-case analyses of reports. In recent years, however, data mining techniques have become more important. The term ‘data mining’ refers to the principle of analysing data from different perspectives and extracting the relevant information. Algorithms are often used to determine hidden patterns of associations or unexpected occurrences i.e. signals in large data-bases. Although the methodology of the various data mining methods applied in pharmacovigilance differ, they all share the characteristic that they express to what extent the number of observed cases differs from the number of expected cases. Several approaches of data mining are currently in use. Proportional reporting ratios (PPRs), compare the proportion of reports for a specific ADR reported for a drug with the proportion for that ADR in all other drugs. The calculation is analogous to that of relative risk. Using the same information, it is also possible to calculate a ‘reporting odds ratio’. The Bayesian confidence propagation neural network (BCPNN) method is used to highlight dependencies in a data set. This approach uses Bayesian statistics implemented in a neural network architecture to analyse all reported ADR combinations.
During the late 1970s and early 1980s, a novel type of active New Zealand developed surveillance (the Intensive Medicines Monitoring Programme) and the United Kingdom (Prescription Monitoring events). These rigorous monitoring systems make use of prescription information to identify drug users.The medication's prescriber is questioned about any side effects taking place while the drug under observation is being used. These to find new signals, data are gathered and examined. The these intensive monitoring systems' methodology has been thoroughly discussed elsewhere . Intensive monitoring is based on a non-interventional observational cohort, setting it apart from spon simultaneous reporting, as the former merely keeps an eye on chosen medications over a specific time frame. Through its with its non-interventional nature, close observation offers actual clinical data that doesn't involve inclusion or criteria for exclusion during the time of collection. It is independent of the type of criteria used for selection and exclusion that define clinical trials, removing the need for selection prejudice. The methodology's foundation is another advantage upon event monitoring and, as a result, can recognize Finding signs of events that weren't necessarily suspected as the medication under study's adverse drug reactions. Intense Programs for monitoring also make it possible for the occurrence of adverse events to be calculated, allowing for quantification of the possibility of specific ADRs.
A study must be conducted in order to test a hypothesis. The research can be carried out in a number of ways, including cohort studies and case-control studies. The Power considerations are one of these methods' limitations. and research methodology. For the purpose of conducting retro comprehensive case-control and cohort studies, information that has been gathered in a dependable and consistent manner must be accessible. The GPRD, or General Practice Research Database as well as the PHARMO Record Linkage System, which will explained in more detail in the sections that follow, were selected because they stand for two distinct kinds of databases from Europe. Additional database and record linking Systems are accessible for study in both Europe and as well as in North America .
Early in the 1990s, the PHARMO record-linking system was created in the Netherlands. Links to PHARMO hospital and community pharmacy data within a particular region based on the patient's gender, birthdate, and GP code. Drug-dispensing records are now part of the system from hospital discharge records and neighborhood pharmacies of roughly two million Dutch citizens. The information the collection dates back to 1987 and is longitudinal. Additional PHARMO has recently been connected to additional data, including as population surveys, primary care data, laboratory and genetic information, accident and cancer databases, and mortality statistics and financial results. The system has clearly defined denominator data that permits prevalence and lence estimates and is reasonably priced due to the fact that Databases are linked and utilized. The database PHARMO is utilized for case-control studies, follow-up research, and other analytical epidemiological research to assess medication effects that are induced. The database has previously been utilized for research on drug use, treatment compliance, ADRs and economic impact .
ADVERSE DRUG REACTION IN PHARMACOVIGILANCE -
The World Health Organization defines pharmacovigilance as "the science and activities related to the detection, assessment, understanding, and prevention of adverse effects or any other drug-related problems."
According to this framework, one of the most important issues in contemporary healthcare is adverse drug reactions, or ADRs. Unintentional, detrimental reactions to drugs that happen at typical therapeutic dosages are known as adverse drug reactions (ADRs), and they are a significant global cause of morbidity, mortality, and financial burden.
The severity of ADRs is demonstrated by recent worldwide data. To illustrate the scope of the issue, pharmacovigilance databases like the FDA Adverse Event Reporting System reported nearly 175,000 deaths and over 1.25 million serious adverse events in a single year.
Classification of Adverse Drug Reaction –
Table no 1 : Frequency of occurrence based on the type of adverse drug reaction
|
Sr no |
Category |
Frequency |
Example |
|
1. |
Very common |
>10% |
Dizziness, fatigue, tiredness. |
|
2. |
Common (frequent) |
>1% and<10% |
Sedation, memory problems, Depression. |
|
3. |
Uncommon (infrequent) |
>0.1% and<1% |
Skin rash |
|
4. |
Rare |
>0.01%and<0.1% |
Stevens Johnson syndrome (Sulphonamide). |
|
5. |
Very rare |
<0.01% |
Aplastic anemia (phenytoin). |
Types Of Adverse Drug Reactions –
Table no 2 : Types of adverse drug reaction
|
Sr no |
Type |
Description |
Examples |
|
1. |
TypeA (Augmented) |
Predicted, Dose dependent, severity increases with increase in dose. |
Predicted, Dose dependent, severity increases with increase in dose. |
|
2. |
TypeB (Bizarre) |
Unpredictable, rare, idiosyncratic, mechanisms are unknown, unrelated to the dose. |
Hepatitis caused by halothane & aplastic anaemia caused by chloramphenicol |
|
3. |
TypeC (Continuous drug use) |
Irreversible,unexpected, unpredictable. |
Dementia by anticholinergic medications . |
|
4. |
TypeD (Delayed) |
Delayed occurrence of ADRs. |
Ophthalmopathy after chloroquine. |
|
5. |
TypeE (End of Dose) |
Withdrawal reactions. |
Seizures on alcohol or benzodiazepines withdrawal. |
|
6. |
TypeF (Failureof therapy) |
Therapeutic failure of drug. |
Accelerated hypertension because of improper therapy . |
CLINICAL TRIALS –
Introduction to Pre-Clinical Trials
Because preclinical research assesses a target compound's safety, efficacy, and potential side effects, it is crucial to the process of discovering and creating new medications. intervention before any testing on humans. These studies help researchers make informed decisions about the progress of clinical trials, allowing for the development of innovative and effective therapies that can improve patients' lives everywhere. In the preclinical phase, laboratory-based tests and experiments are typically conducted using either in vivo or in vitro testing.
Preclinical study types include:
1. In vitro studies: which are experiments carried out in lab settings using organs, tissues, or cells (such as "organ-on-a-chip" systems or cell cultures).
2. In Silico Research: Using computer simulations and modeling to forecast how a drug may act in the human body, improving research design and minimizing animal testing.
3. In Vivo Research: Examining living things, mainly animal models usually two mammalian species—one rodent and one non-rodent, like dogs and mice to evaluate the effects of a medication on an entire biological system .
Introduction of Clinical Trials -
Overview of Clinical Trials: A clinical trial is a methodical procedure designed to determine the effectiveness and safety of a medication or tool for diagnosing, treating, or preventing a medical condition. Clinical investigation comprises several stages, such as phase 0 (micro-dosing studies), phase 1, phase 2, phase 3, and phase 4. Phases 0 and 2 are referred to as exploratory trial phases, while phase 1 is known as the non- Phase 3 is referred to as the therapeutic confirmatory phase, Phase 4 is called the post-approval or post-marketing surveillance stage. Clinical trials are a particular type of clinical research intended to assess the effectiveness and safety of medical interventions, such as medications, equipment, therapies, and preventative measures.
Phases of Clinical Trials -
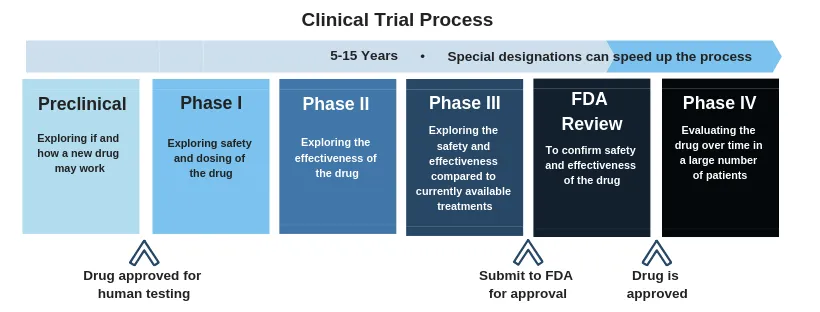
Fig no 2: Process of clinical trials
Regulatory bodies that approves Clinical Trials –
These agencies ensure that the trial meets scientific, safety, and quality standards. Examples:
ROLE OF HEALTHCARE PROFESSIONALS IN PHARMACOVIGILANCE -
Clinicians' role in drug safety and pharmacovigilance:
By identifying, treating, and reporting adverse drug reactions (ADRs) to national pharmacovigilance centers (NPCs), clinicians play a critical role in preventing ADRs. Therapeutic reasoning and the right drug selection for each patient are necessary for the safe and sensible prescription of medications . Age, medication errors, polypharmacy, and patient-specific risk factors like comorbidities can all raise the risk of adverse drug reactions (ADRs).
Roles of national pharmacovigilance centre
Coordinating national ADR monitoring programs is NPCs' primary responsibility. This typically entails keeping an eye on, looking into, and evaluating ADR reports that are obtained from medical professionals and product license holders .
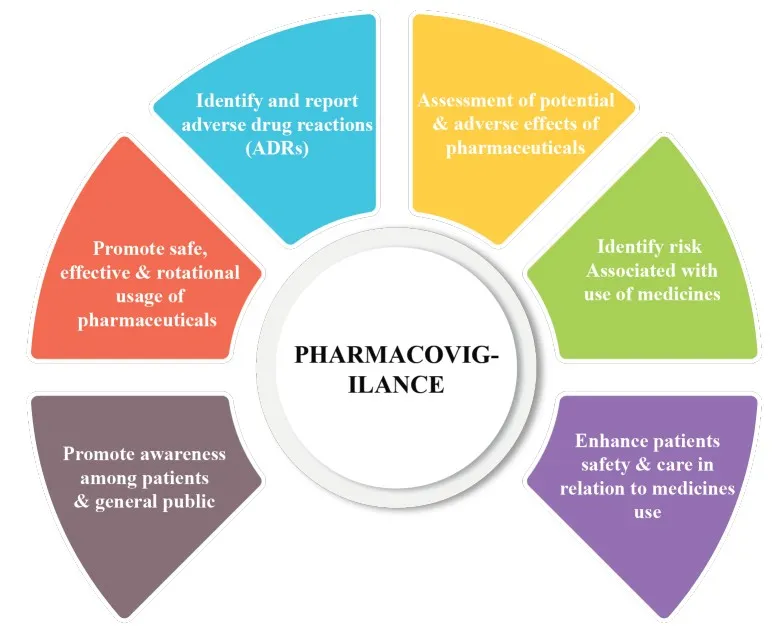
Fig No 3 : Role of Pharmacovigilance.
Product license holders are accountable for their products on the market, so they must report all pertinent safety information and adhere to post-marketing regulations. This includes responding quickly to requests for data needed to perform a benefit-risk analysis so that the proper regulatory measures can be implemented. The Council for International Organizations of Medical Sciences (CIOMS) and the International Conference on Harmonization (ICH) offer scientific recommendations for pharmacovigilance and risk management of pharmaceuticals at every stage of their life cycle, from preclinical and clinical development to post-marketing . Pharmaceutical companies can report suspected adverse drug reactions (ADRs) using the CIOMS form, which follows a standard reporting procedure that has been proven to be beneficial and efficient .
In addition to handling product defect reports, some NPCs may have portfolios that include cosmetics and personal hygiene items. For instance, the Center for Food Safety and Applied Nutrition's Adverse Event Reporting System, a public database of adverse events pertaining to foods, dietary supplements, and cosmetics, was established by the US Food and Drug Administration in 2016 .
CROSSROADS OF ANIMAL MEDICINES ( VETERINARY PHARMACOVIGILANCE ) -
With mounting evidence that veterinary medications are being used recreationally and in illegal drug markets, their abuse has become a major public health concern. This study used a variety of techniques, such as social media analysis, pharmacovigilance data approaches, and a systematic literature review, to look into veterinary medication abuse.
Techniques: 28 veterinary medications, mainly α-2 and β-2 adrenergic receptor agonists, GABAergic modulators, opioid receptor agonists, NSAIDs, and NMDA receptor antagonists, were found to be abused by humans in a systematic review of 66 articles. 38,756 adverse events were retrieved from a pharmacovigilance analysis of 21 veterinary medications using the FAERS database. Themes associated with misuse trends were examined in a netnographic dual-method analysis of Reddit discussions.
Outcomes: Veterinary medications were abused for recreational purposes, pain management, bodybuilding, weight loss, and stress-related self-medication. Inhalation, oral, and parenteral administration were common methods. Affordability, accessibility, and the simplicity of getting several prescriptions were among the driving forces. Levamisole, pentobarbital, and xylazine were among the most dangerous drugs, according to FAERS analysis, which showed 9566 fatalities. Ninety percent of cases showed signs of polysubstance use. Emerging abuse of xylazine, carfentanil, medetomidine, pentobarbital, phenylbutazone, and acepromazine was discovered through netnography.
In order to address the risks of overdose, dependence, and illicit drug adulteration, this study highlights the growing misuse of veterinary medications and the necessity of increased vigilance in healthcare and public health policy.
CASE STUDY : SAFETY AND REACTOGENICITY OF BNT162b2 COVID-19 VACCINE –
A substantial amount of safety data was produced by the clinical development programs for coronavirus disease 2019 (COVID-19) vaccines, including BNT162b2, the subject of this review. Strong efficacy and short-term safety data (up to two months of follow-up post-primary schedule for BNT162b2, in accordance with regulatory guidance) served as the foundation for the initial approvals. These approvals were modified to include a booster dose based on a 2.6-month follow-up after the BNT162b2 booster. Six Following approval, vaccine producers carried out clinical trials and worked with regulatory bodies and other groups to monitor longer-term safety through post-marketing pharmacovigilance initiatives. This post-authorization safety data soon outpaced the amount and scope of data produced by clinical trials. In addition to independent clinical and real-world trials in special populations and with various vaccination regimens, which validated the safety profile seen in clinical trials, permitted analysis of real-world practice patterns, and produced an unprecedented level of transparency, this was supported by real-world data from nations starting mass vaccination programs.
As soon as the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) emerged, it became apparent that vaccines were urgently needed to combat the rapidly spreading pandemic. Several stakeholders worked together to accelerate vaccine development, authorization, and roll-out while upholding strict clinical trial standards.
The use of novel vaccine technologies with shorter design-to-production times, increased funding and collaboration, at-risk investment in commercial production from manufacturers prior to approval, accelerated regulatory procedures, and accelerated review timelines all contributed to the compression of vaccine clinical development timelines. Additionally, compared to typical vaccine development timelines, high infection rates and participant willingness to participate in clinical trials resulted in faster study completion and enrollment. Rapid production and scale-up of messenger RNA (mRNA) vaccines were made possible by pre-existing manufacturing processes. 1 As a result, compared to other vaccines, like influenza, development and commercialization timelines were significantly shorter, and all pathway steps necessary for regulatory approval were completed.
As booster doses and variant-adapted bivalent vaccines have been created and introduced to the market, the continuous emergence of new SARS-CoV-2 variants has prompted additional generation of safety data for mRNA vaccines.
The US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) both committed additional resources to facilitate the quick development and approval of vaccines. 7 While requiring manufacturers to provide additional vaccine data from clinical settings for thorough review, the FDA EUA pathway made COVID-19 vaccines rapidly accessible to the public in an emergency pandemic setting. 7 In a similar vein, the EMA accelerated its evidence appraisal procedure by implementing rolling review stages, which enabled regulators to obtain and examine the data as soon as they were made available. In order to accelerate deadlines while maintaining the development process, regular communication between manufacturers, regulatory bodies, and other stakeholders was crucial. Regulators were able to evaluate marketing authorization applications more quickly thanks to increased familiarity with the data and prearranged opportunities for sponsor/regulator conversations. 3 On December 21, 2020, the European Commission initially authorized BNT162b2 for conditional marketing.
Development in children
A clinical trial was conducted with participants aged 6 months to 11 years to evaluate pediatric age-related doses of BNT162b2 after the vaccine was approved for adult populations. Two doses of BNT162b2 (10 μg, 20 μg, or 30 μg) were given to the cohort of children aged 5 to 11 in the Phase I section (NCT04816643). The 10 μg dose was chosen for additional evaluation in the Phase II/III section due to a higher frequency of fever with the higher doses and because the neutralizing antibody profiles of the 10 μg and 20 μg doses were similar. 36. Participants in the cohort of children aged 6 months to 4 years were given either 10 μg or 3 μg of BNT162b2. The 3 μg dose was chosen for additional evaluation due to a higher frequency and more severe reactogenicity to the 10 μg dose compared to the 3 μg dose, as well as neutralizing antibody profiles across dose levels that were comparable to those seen in older age groups.
The majority of local and systemic reactions in a cohort of 3,013 children aged 6 months to 4 years who received BNT162b2 were mild to moderate in intensity, and there were no reports of Grade 4 local reactions. There were no fatalities, the frequency of adverse events (AEs) was comparable between BNT162b2 and placebo recipients, and few participants were discontinued as a result of AEs. 37.
ROLE OF PHARMACOVIGILANCE IN INDIA –
December 1961 saw the official introduction of pharmacovigilance (PV) with the release of a letter (case report) published in the Lancet by Australian physician W. McBride, who initially suspected a causal connection between thalidomide, a medication used during pregnancy, and severe fetal abnormalities (phocomelia) Pregnancy: Thalidomide was administered to expectant mothers as a sedative and antiemetic. Within the World Health Organization (WHO) advertised the "Programme for International Drug A pilot project called "Monitoring" sought to centralize global data on adverse drug reactions (ADRs). Within Specifically, the "WHO Programme's" primary goal was to determine the earliest potential PV signals. A group of French pharmacologists coined the term PV in the middle of the 1970s.Toxicologists should specify the actions that support "The assessment of the risks of side effects."possibly connected to medication therapy . PV is the science of gathering, tracking, investigating, evaluating, and assessing data from medical professionals and patients regarding the negative consequences of drugs, biological products, blood products, herbal remedies, vaccines, medical equipment, and complementary and traditional medicines with a view to discovering fresh data regarding product-related risks and avoiding injury to patients. The difficulty of preserving public trust while optimizing drug safety has grown more intricate. Biotechnology and pharmaceutical firms must not only monitor, as well as proactively assess and control medication risk over the course of a product's lifecycle, from creation to the post-market
Scope of pharmacovigilance in india :
Since the 1972 WHO technical report, the field of PV has advanced significantly, and it continues to be a vibrant scientific and clinical field. Overcoming the obstacles has been crucial of the growing variety and effectiveness of pharmaceutical and biological medications, such as vaccines, which have an unavoidable and occasionally unpredictable risk of side effects. However, there is a lower chance of injury when medications are administered by a qualified medical professional and by patients who are aware of and accountable for their medications. When unfavorable effects and toxicity manifest, especially when previously unidentified in connection with the medicine, it is crucial that they are evaluated and effectively conveyed to a group of people who possesses the expertise to understand the data This is PV's function, which has already been accomplished. However, more is needed to incorporate the field into clinical practice and public policy.
Fig No 4 : A typical pharmacovigilance setup
Framework of New Program
The new program's framework the IPC center is dedicated to creating India's own drug information and adverse drug reaction database so that India won't need to rely on information from other nations to make decisions about to the suspension and prohibition of drugs. India currently lacks a robust database on ADRs and must rely on data from Western nations. Thus far, just 2823 ADRs have been documented since September 2010 under the current PVPI, which is insufficient to generate any significant conclusion associated with a specific signal.
Table No 3: Chronological Developments in PV
It is anticipated that all hospitals, clinics, educational institutions, and public health programs across the nation, both public and private, will participate in the PVPI and report ADRs to IPC so that all generated data can be gathered and examined in one location. The PV field's historical developments, with particular reference to India, are listed in Table 6. Figure 10 displays the PVPI-managed governing body and monitored centers.
Fig 5 : Pharmacovigilance program in India and responsibilities.
Three stages were planned for the program's implementation. 40 ADR would be included in phase I 2010 will see the implementation of monitoring centers (AMC). Phase II of the program would be expanded to incorporate up to 140 medical schools accredited by MCI by 2011. Up until 2011's conclusion, a total of there are just 60 AMCs included. In the end, Phase III would encompass the whole healthcare system by 2013 [62]. The corresponding zonal CDSCO provides operational and logistical support to the AMCs centers located in Chennai, Mumbai, Kolkata, and Ghaziabad. The CDSCO zonal centers will be under the administrative jurisdiction of the New Delhi-based CDSCO headquarters. Structure of organizations of PVPI and their corresponding duties are displayed in Figure.
Role of Artificial Intelligence –
A new era is being ushered in by artificial intelligence (AI). Unbeknownst to us, technology has permeated every aspect of our daily lives, from the house to the street, and is currently influencing pharmacovigilance (PV), scientific research, and the healthcare system. By analyzing suspected adverse reaction reports and extracting health data to find drug safety signals, PV aims to lower the incidence and risk of medication use as soon as possible. Individual case safety reports (ICSRs) have been used to gather postmarketing safety reports for medical products globally through a spontaneous reporting system in an organized and methodical manner. Although they contain unstructured text, electronic health records, regular safety update reports, published medical literature, registries, and pharmacoepidemiology studies are supplementary data sources for routine PV practices.
Need of Artificial Intelligence –
The number of suspected adverse event (AE) reports in the PV database has increased exponentially [Figure 1]. For important stakeholders like pharmaceutical companies, regulatory bodies, medical and PV specialists, and managers of National Pharmacovigilance Programs, processing the massive volume and variety of data sources, making sensible use of it, and separating "needles from haystack" is a challenge. Essential components (patient, reporter, adverse reaction, suspected and concurrent medications, and outcome) are typically required for ICSR case processing. ICSR case processing is also assessed for the likelihood of a causal relationship, the expectedness of adverse events (AEs) according to the prescribing information leaflet, the severity and seriousness criteria, and, lastly, for completeness and validity for regulatory submission. Crucially, it includes both human cognition and manual tasks [Figure 2]. In essence, it is costly and time-consuming because it requires labor and technical know-how. There has been a lot of excitement and enthusiasm to use AI technology to automate PV in order to deal with this increased workload.
Opportunity and benefits –
It has been suggested that the AI tool will help with routine and repetitive manual tasks like data entry, AE identification, drug-drug interactions, subtle data patterns, and single case review. AI can also transform handwritten documents and unstructured, free-text drug safety data into a machine-readable format.
Additionally, the tool can identify serious reports and exclude nonserious reports, check duplicate reports, classify reports into physician or consumer reports, and automate the Medical Dictionary for Regulatory Activities coding. It's interesting to note that the AI platform can also analyze unstructured data, extract text, and find pertinent information to create clinically sound auto-narratives and spot patterns in both structured and unstructured narratives. This eliminates the need for manual signal identification and validation as well as routine review of individual cases. Additionally, it can extract ICSR data from a variety of published documents, including discharge summaries, free-text clinical notes in electronic health records, case reports, medication reviews on social media, and medical literature. According to a recent survey, scientists can save time and money by using AI tools to process data quickly and perform calculations that were previously impractical. Adoption of AI tools will lower the effort, time, and cost of case processing, improve data quality, and potentially revolutionize PV activities due to the vast amount of drug safety data that is stored electronically
Challengees of adoption of AI in pharmacovigilance.
Despite being a promising tool, its implementation and practical impact raise several questionsZand challenges [Table 1]. Whether the qualitative assessment by AI algorithm to determine causality and safety signals (that requires clinical evaluation plus expert opinion) be as good as human experts for decision making? Whether the AI algorithm can be integrated across the PV lifecycle to identify black swans? Will it completely replace PV professionals? There seem to be no simple straightforward answers. Let us introspect them from PV case processing and application perspectives in the contex health-care systems.
DISCUSSION
By tracking, identifying, evaluating, and averting adverse drug reactions (ADRs), pharmacovigilance plays a significant part in guaranteeing medication safety and enhancing patient care. The significance of pharmacovigilance in healthcare systems and the pharmaceutical industry is emphasized in this project on "Pharmacovigilance in the Modern Era." The study describes how ADR monitoring enhances therapeutic outcomes and reduces medication-related risks. Strong pharmacovigilance systems are essential for public safety, as evidenced by historical occurrences like the thalidomide tragedy and the withdrawal of rofecoxib (Vioxx).
The project also covers the function of ADR reporting systems and the role that patients, pharmaceutical companies, regulatory bodies, and medical professionals play in preserving drug safety. Individual Case Safety Reports (ICSRs) and spontaneous reporting systems continue to be the cornerstone of pharmacovigilance efforts globally. However, traditional manual methods are becoming time-consuming and challenging to manage effectively due to the rapid increase in safety data.
The incorporation of artificial intelligence (AI) into pharmacovigilance is a significant feature of this project. Large amounts of safety data can be processed, adverse events can be found, duplicate reports can be found, coding systems can be automated, and unstructured medical data can be analyzed more effectively with AI technologies. AI tools have the potential to greatly lessen workload, save time, increase accuracy, and facilitate early signal detection.
Despite these benefits, there are drawbacks to using AI in pharmacovigilance. Clinical judgment, determining causality, and making final decisions still require human expertise. Because safety evaluation frequently necessitates medical knowledge, ethical consideration, and expert interpretation, AI cannot fully replace pharmacovigilance professionals. Therefore, for pharmacovigilance practice to be effective, a combined strategy involving both cutting-edge technology and qualified professionals is required.
Overall, this project concludes that as technology advances, pharmacovigilance is constantly changing. Drug safety monitoring could be strengthened and public health outcomes could be improved by using contemporary tools like artificial intelligence (AI) and digital healthcare systems. Future pharmacovigilance growth depends on ongoing awareness, accurate ADR reporting, regulatory support, and technological innovation.
CONCLUSION
A crucial part of the healthcare system that guarantees the effectiveness, safety, and appropriate use of medications is pharmacovigilance. It is essential for the identification, evaluation, tracking, and avoidance of adverse drug reactions (ADRs) and other drug-related issues. The development of pharmacovigilance has greatly enhanced patient safety and assisted regulatory bodies in making prompt decisions about medication use. Drug safety incidents in the past have shown how crucial it is to keep an eye on medications even after they have been approved and marketed.
Traditional pharmacovigilance systems now face new difficulties due to the growing amount of healthcare and medication safety data. Pharmacovigilance procedures have changed as a result of the introduction of cutting-edge technologies like artificial intelligence (AI), machine learning, electronic health records, and automated reporting systems. Faster signal detection, effective case processing, better data management, and improved identification of possible safety issues are all made possible by these technologies. AI has demonstrated significant promise in lowering manual labor and improving the precision and effectiveness of pharmacovigilance operations.
However, human expertise is still necessary for clinical evaluation, causality assessment, and regulatory decision-making despite technological advancements. Effective pharmacovigilance requires cooperation among healthcare professionals, pharmaceutical industries, regulatory authorities, and patients to ensure accurate reporting and monitoring of ADRs.
To sum up, pharmacovigilance is constantly changing to satisfy the needs of contemporary healthcare. Strong regulatory frameworks, cutting-edge technology, and engaged healthcare stakeholders can all work together to improve public health globally and bolster drug safety monitoring.
REFERENCES






Shrushti Krishnani, Tejaswini Patil, Vaibhavi Kate, Rakesh Vasave, Hemant Nikumbh, Rahul Patil, Pharmacovigilance In the Modern Era, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 5, 7652-7671, https://doi.org/10.5281/zenodo.20426321
 10.5281/zenodo.20426321
10.5281/zenodo.20426321