
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Vidya Niketan Institute of Pharmacy and Research Center, Bota, Sangamner, Ahilyanagar 422602
Quantitative Structure–Activity Relationship (QSAR) has emerged as one of the most powerful computational approaches in rational drug design, bridging the gap between chemical structure and biological activity. By utilizing molecular descriptors such as hydrophobicity, electronic distribution, and steric parameters, QSAR enables researchers to build predictive models that correlate structural features with pharmacological responses. The significance of QSAR lies in its ability to reduce experimental costs, shorten timelines, and minimize animal testing by identifying and optimizing lead compounds in silico before synthesis. Over the decades, QSAR has evolved from simple linear regression models into sophisticated multidimensional approaches, including 2D, 3D, and even 6D-QSAR, which integrate receptor interactions, conformational flexibility, and machine learning algorithms. In drug discovery, QSAR is widely applied in lead identification, optimization, and toxicity prediction. It has been successfully used in the development of cardiovascular drugs, anticancer agents, antivirals, and antibiotics.
Drug discovery is a complex, expensive, and time-consuming process that has remained one of the greatest challenges in modern pharmaceutical sciences. The traditional pipeline, starting from the identification of bioactive compounds through clinical evaluation, can take more than a decade and require investments exceeding billions of dollars. Despite such massive investments, the success rate of new drug candidates remains discouragingly low. Studies suggest that only one out of 5,000–10,000 screened compounds eventually reaches the market, with many candidates failing due to poor efficacy, toxicity, or unfavorable pharmacokinetic profiles. This high rate of attrition not only increases costs but also delays the delivery of novel therapies to patients who are in urgent need.[1]
To address these challenges, the pharmaceutical industry has progressively turned toward computational approaches to streamline drug discovery. Over the last few decades, in silico methodologies have evolved from basic molecular modeling techniques to highly sophisticated computer-aided drug design (CADD) tools. These strategies enable researchers to simulate drug–target interactions, predict biological activities, and evaluate pharmacokinetic and toxicological properties prior to laboratory synthesis. Among these computational strategies, Quantitative Structure–Activity Relationship (QSAR) modeling stands out as one of the most widely used and reliable tools in rational drug design.[2]
QSAR is based on the fundamental principle that the biological activity of a compound is directly related to its chemical structure. By mathematically correlating molecular descriptors—such as hydrophobicity, electronic properties, steric parameters, and hydrogen-bonding capacity—with observed biological responses, QSAR provides predictive insights into the activity of novel compounds. This approach allows medicinal chemists to identify lead molecules, optimize drug-like properties, and minimize toxicity risks without extensive experimental screening. The integration of QSAR into the early stages of drug design significantly reduces both cost and time, making the process more efficient and sustainable.[3]
The importance of QSAR extends beyond predicting activity. It plays a pivotal role in addressing several critical aspects of drug discovery, including:
Lead Identification and Optimization: Assisting in the selection of promising compounds from large chemical libraries.
ADMET Prediction: Evaluating absorption, distribution, metabolism, excretion, and toxicity profiles before clinical testing.
Drug Repurposing: Identifying new therapeutic indications for existing drugs.
Environmental and Regulatory Sciences: Assessing the toxicity of chemicals and aiding in regulatory decision-making.[5]
Given its wide applications, QSAR has undergone significant evolution since its inception in the 1960s with Hansch and Fujita’s linear models. The field has progressed from simple regression-based 2D models to advanced multidimensional approaches, including 3D-QSAR, 4D-QSAR, and even 6D-QSAR, integrating machine learning and artificial intelligence for enhanced accuracy.
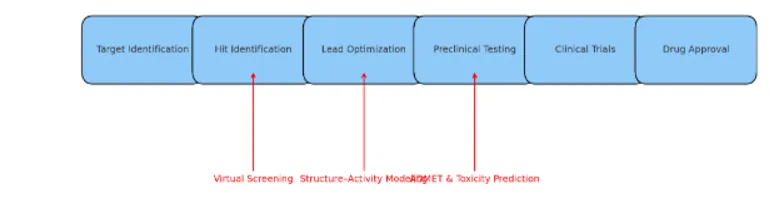
Figure 1 : Role of QSAR in the Drug Discovery Pipeline
Objective of this Review
The purpose of this review is to provide a comprehensive overview of QSAR and its role in modern drug design. Specifically, this paper will:
By systematically exploring these aspects, this review aims to highlight the transformative role of QSAR in reducing the time, cost, and risks associated with drug discovery, ultimately supporting the development of safer and more effective therapeutic agents.
Historical Background of QSAR
The concept of relating chemical structure to biological activity has been a cornerstone of medicinal chemistry for more than a century. However, the systematic and quantitative framework that underpins modern QSAR (Quantitative Structure–Activity Relationship) emerged in the 1960s through the pioneering work of Corwin Hansch. His contributions laid the foundation for computational drug design and still form the basis of many QSAR studies today.
The Work of Corwin Hansch and the Birth of QSAR
In the early 1960s, Corwin Hansch, a professor of chemistry at Pomona College, recognized that the biological activity of a compound was influenced by multiple physicochemical properties, such as lipophilicity, electronic distribution, and steric effects. He proposed a systematic method to mathematically correlate these properties with biological responses. This idea became the foundation of the QSAR field.
The classical Hansch–Fujita equation (1964) expressed biological activity (log 1/C, where C = molar concentration producing activity) as a linear function of molecular descriptors:
log(1/C) = aπ + bσ + cE? + k
Where:
This equation provided the first formal mathematical model linking structure to activity, enabling researchers to predict biological effects of untested compounds. The Hansch–Fujita approach was revolutionary because it offered not only mechanistic insights but also predictive power.
Evolution and Milestones in QSAR Development
Since Hansch’s work, QSAR has undergone continuous refinement and expansion, leading to multiple “generations” of models:
These milestones illustrate how QSAR evolved from simple linear models to multidimensional frameworks that incorporate advanced computational and biophysical concepts.[7]
Impact of Computational Advancements
The evolution of QSAR has been closely tied to progress in computational technologies. Early QSAR models were restricted to small datasets due to limited computing power. With the advent of high-performance computing, molecular modeling software, and machine learning algorithms, the scope of QSAR has dramatically expanded. Today, researchers can analyze large chemical libraries containing millions of compounds and employ sophisticated nonlinear methods such as:
Artificial Neural Networks (ANNs)
Support Vector Machines (SVMs)
Random Forests (RF)
Deep Learning (DL)
These advancements have enhanced predictive accuracy and applicability, transforming QSAR into a central tool of modern drug discovery. Additionally, integration with databases (e.g., PubChem, ChEMBL) and big data approaches has allowed QSAR to reach beyond drug design, impacting toxicology, regulatory sciences, and environmental chemistry.[11]
Fundamentals of QSAR
Definition and Concept
Quantitative Structure–Activity Relationship (QSAR) is a computational modeling technique that establishes a mathematical correlation between the structural properties of chemical compounds and their biological activity or physicochemical behavior. The fundamental assumption of QSAR is the similarity principle: “structurally similar molecules exhibit similar biological activities.”
In drug discovery, QSAR is used to predict the activity of untested compounds, optimize lead molecules, and reduce the need for expensive experimental screening. QSAR integrates chemistry, biology, mathematics, and statistics to provide predictive insights that accelerate rational drug design.[13]
Stepwise QSAR Approach
Data Selection
Descriptor Calculation
Model Building
Validation
Prediction
General QSAR Equation
The classical Hansch–Fujita type QSAR equation can be represented as:
Biological Activity=a(Hydrophobic)+b(Electronic)+c(Steric)+d
log(1/C?)=aπ+bσ+cEs?+k
Where:
Molecular Descriptors in QSAR
Molecular descriptors form the foundation of QSAR modeling, as they provide a quantitative representation of chemical structures. They transform complex molecular information into numerical values that can be correlated with biological activity. These descriptors capture essential physicochemical, structural, and electronic features of compounds, thereby helping in the prediction of pharmacological profiles. Based on their nature, molecular descriptors can be broadly categorized into hydrophobic, electronic, steric, and advanced descriptors.[29]
Hydrophobicity Descriptors
Hydrophobic interactions play a central role in determining drug–receptor binding affinity, membrane permeability, and bioavailability. Hydrophobic descriptors measure the lipophilic character of molecules.
Key Parameters
Relevance to Lipinski’s Rule of Five
Electronic Descriptors
Electronic effects influence how drugs interact with their biological targets, particularly in terms of binding strength, charge distribution, and reactivity.
Key Parameters
Relation to Receptor Binding
5.3 Steric Descriptors
Steric factors describe the three-dimensional shape, size, and spatial distribution of substituents. Since biological targets have specific binding pockets, steric compatibility is essential for activity.
Key Parameters
Example
Advanced Molecular Descriptors
Beyond basic hydrophobic, electronic, and steric descriptors, modern QSAR employs advanced descriptors derived from computational chemistry and topology.
1. Topological Indices
2. Quantum Chemical Parameters
3. Molecular Field Descriptors (3D-QSAR)
Types of QSAR Models
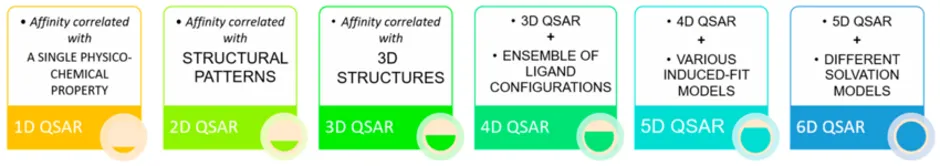
QSAR models have gradually advanced in their dimensionality to capture more complex molecular and biological information. From simple physicochemical correlations (1D) to sophisticated multidimensional approaches integrating receptor interactions and dynamics (6D), these models demonstrate the progressive refinement of computational drug design.
One-Dimensional (1D-QSAR)
Two-Dimensional (2D-QSAR)
Three-Dimensional (3D-QSAR)
Four-Dimensional (4D-QSAR)
Five-Dimensional (5D-QSAR)
Six-Dimensional (6D-QSAR)
Statistical & Computational Methods in QSAR
The success of Quantitative Structure–Activity Relationship (QSAR) studies largely depends on the statistical and computational tools used to correlate molecular descriptors with biological activities. Since chemical and biological data are often multidimensional and complex, advanced statistical methods and machine learning approaches are necessary to extract meaningful insights. This section outlines the major computational strategies and validation techniques used in QSAR modeling.
Regression Analysis
Regression techniques form the backbone of classical QSAR. They aim to establish a mathematical relationship between molecular descriptors (independent variables) and biological activity (dependent variable).
The simplest and most widely used method in QSAR. MLR assumes a linear relationship between descriptors and activity. Its advantages include simplicity and interpretability; however, it may fail in the presence of highly correlated or nonlinear data.
An extension of regression analysis, PLS is particularly effective when the number of descriptors is very large compared to the number of compounds. It reduces dimensionality while maximizing correlation between descriptor space and biological activity, making it a preferred method in 3D-QSAR (e.g., CoMFA and CoMSIA).[31]
Principal Component Analysis (PCA)
PCA is a data reduction method that transforms a large set of correlated descriptors into a smaller set of uncorrelated principal components.
Artificial Neural Networks (ANN)
ANNs are nonlinear statistical learning models inspired by the human brain. They can capture complex, nonlinear relationships between molecular descriptors and biological responses.
Machine Learning & Artificial Intelligence (AI) in QSAR
In recent years, machine learning (ML) and AI have revolutionized QSAR modeling by offering powerful predictive capabilities.
Model Validation Methods
A QSAR model is only as good as its predictive reliability. Hence, validation is a critical step in QSAR studies.
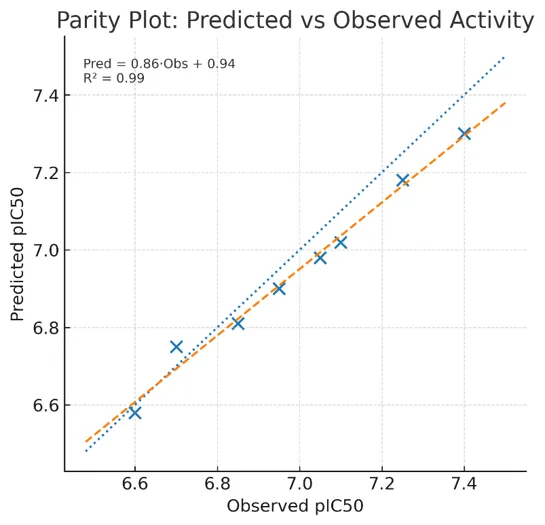
A robust QSAR should meet criteria such as a defined endpoint, unambiguous algorithm, defined domain of applicability, and predictive power.[26]
Applications of QSAR in Drug Design
Drug design is no longer solely dependent on traditional synthesis and experimental testing. With the advent of computational tools, QSAR has emerged as a powerful predictive methodology that saves both time and resources by correlating molecular structure with biological activity. QSAR applications can be broadly classified into lead optimization, virtual screening, toxicity prediction, and therapeutic case studies.
Lead Optimization
QSAR plays a critical role in optimizing lead compounds by guiding chemists on which substituents improve biological activity and which reduce it.
Impact:
Virtual Screening
QSAR models are increasingly applied in virtual screening of chemical libraries to identify drug-like compounds before laboratory synthesis.
Example:
Toxicity Prediction (In Silico Toxicology)
Toxicity is a major cause of drug failure in clinical trials. QSAR models help in early prediction of toxic liabilities.
Example:
Case Studies
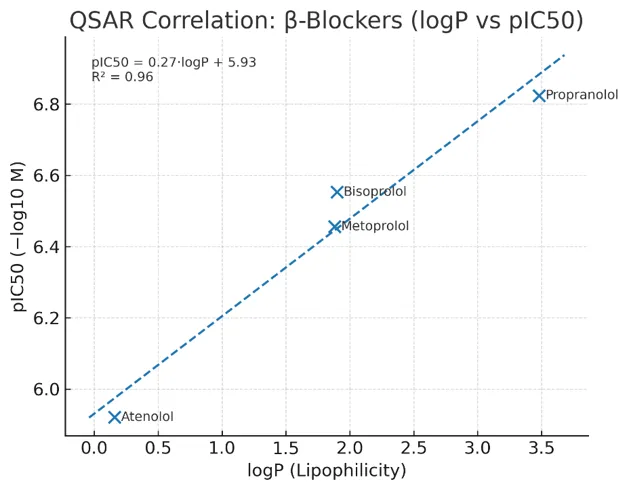
(a) Beta Blockers – Correlation with Lipophilicity
Table No 1
|
Compound
|
logP |
Experimental Activity (IC??) |
Predicted Activity (QSAR) |
|
Propranolol |
3.48 |
0.15 µM |
0.17 µM |
|
Atenolol |
0.16 |
1.2 µM |
1.3 µM |
(b) Sulfonamide Antibacterials – Electronic Effects
Equation Example:
pMIC = 0.62σ + 1.45 (R² = 0.89)
(c) HIV Protease Inhibitors – 3D-QSAR (CoMFA Studies)
(d) Anti-Cancer Agents
Equation Example:
IC?? = -0.35(HOMO) + 0.42(logP) + 1.25 (R² = 0.92)
Advantages of QSAR
Quantitative Structure–Activity Relationship (QSAR) approaches have become indispensable in modern drug discovery and development. By establishing mathematical relationships between chemical structure and biological activity, QSAR offers multiple benefits over conventional experimental methods. The major advantages are summarized below:
1. Time-Saving and Cost-Effective
QSAR reduces the need for synthesizing and testing large numbers of compounds in the laboratory. Once a predictive model is developed, it can screen thousands of virtual molecules in silico within hours, thereby accelerating the lead identification and optimization process. [21]This significantly lowers the overall research and development (R&D) cost compared to traditional high-throughput screening methods.
2. Reduction in Animal Testing
Ethical concerns and regulatory requirements have increased the demand for alternatives to animal testing. QSAR models serve as computational replacements for in vivo studies by predicting pharmacokinetic properties, efficacy, and toxicity profiles before actual synthesis. This minimizes reliance on animal experiments while still ensuring drug safety and effectiveness.
3. Mechanistic Insights
QSAR not only predicts activity but also provides valuable mechanistic insights into drug–receptor interactions. By analyzing the contribution of physicochemical parameters such as hydrophobicity (logP), electronic effects (Hammett σ constants), and steric factors (molecular volume, 3D descriptors), researchers can identify which structural modifications enhance potency or selectivity. This mechanistic understanding guides rational drug design rather than trial-and-error synthesis.[20]
4. High Predictive Power
Well-validated QSAR models demonstrate high predictive accuracy, often with correlation coefficients (R²) above 0.80 and cross-validated Q² values confirming robustness. This predictive power allows medicinal chemists to reliably forecast biological activity, ADME (Absorption, Distribution, Metabolism, Excretion), and toxicological properties, making QSAR a valuable decision-making tool in the early stages of drug design.
5. Integration with Modern Tools
QSAR integrates seamlessly with molecular docking, pharmacophore modeling, and machine learning algorithms. When combined with big data and AI, QSAR achieves higher precision, offering multi-parameter optimization for potency, selectivity, and drug-likeness simultaneously. This multi-dimensional approach enables more effective identification of “drug-like” candidates with fewer experimental iterations.[18]
Limitations of QSAR
While QSAR has proven to be a powerful computational tool in modern drug discovery, it is not without challenges. The accuracy and reliability of QSAR models are highly dependent on the quality of input data, the chosen descriptors, and the statistical methods employed. Several limitations must be acknowledged:[16]
1. Dependence on Quality of Data
The reliability of QSAR predictions is only as strong as the experimental data used to build the model. Inaccurate or incomplete biological activity data, poor assay reproducibility, or errors in chemical structure representation can lead to weak or misleading correlations. Thus, QSAR models require robust, well-curated datasets to achieve high predictive accuracy.
2. Limited Chemical Space
QSAR models are often derived from a restricted set of structurally similar compounds. As a result, their predictive power decreases when applied to chemical scaffolds outside the training set. This limitation reduces the generalizability of QSAR models and poses challenges in exploring novel chemical spaces for first-in-class drug discovery.
3. Overfitting Issues
Overfitting occurs when a model is too complex, capturing noise rather than meaningful trends in the data. Although such models may perform well with the training dataset, they fail to predict accurately for external or unseen compounds. Overfitting remains a common issue, particularly when models are built with numerous descriptors and small datasets.[15]
4. Poor Applicability for Flexible Molecules
QSAR models struggle to predict biological activity for highly flexible molecules with multiple conformations. Since QSAR typically relies on static descriptors (1D–2D), it does not always account for conformational dynamics or receptor-induced fit, leading to reduced accuracy. Although 3D- and 4D-QSAR approaches attempt to address this limitation, challenges remain for compounds with significant flexibility.
5. Simplified Representation of Complex Biology
QSAR assumes that chemical structure alone determines biological activity. However, real biological systems involve complex processes such as protein conformational changes, signaling pathways, and metabolic transformations that cannot always be captured by QSAR descriptors. This simplification sometimes leads to gaps between in silico predictions and in vivo results.
Future Perspectives of QSAR
The field of QSAR has advanced tremendously from simple linear equations to sophisticated multidimensional models. However, the future of QSAR lies in its ability to integrate with cutting-edge technologies and harness the power of big data to address the challenges of modern drug discovery. The following directions highlight how QSAR will evolve in the coming years:[14]
1. Integration with Artificial Intelligence, Machine Learning, and Deep Learning
The advent of AI and deep learning algorithms has revolutionized computational drug discovery. Unlike traditional QSAR models that rely on linear or nonlinear regression, AI can capture hidden and complex nonlinear patterns in large datasets. Machine learning algorithms such as Random Forests, Support Vector Machines (SVM), and Gradient Boosting are increasingly applied to QSAR, enhancing prediction accuracy. Deep learning, particularly neural networks with multiple hidden layers, can process vast descriptor sets and provide predictive models with high generalizability. Integration of QSAR with AI is expected to minimize overfitting and improve the prediction of activities for novel, structurally diverse compounds.
2. Role of Big Data in QSAR
The exponential growth of chemical and biological data from high-throughput screening (HTS), combinatorial chemistry, and omics technologies has created an unprecedented opportunity for QSAR. The future of QSAR will depend on big data analytics that can manage, filter, and interpret massive datasets. Access to global chemical repositories such as PubChem, ChEMBL, and ZINC, combined with advanced computational power, will allow the construction of highly robust, data-driven QSAR models. These models can extend across diverse chemical spaces, thereby improving their predictive applicability.[12]
3. Multi-Target QSAR for Polypharmacology
Traditional QSAR models generally focus on single-target predictions. However, modern drug discovery increasingly recognizes the importance of polypharmacology, where one drug interacts with multiple biological targets. Multi-target QSAR (mt-QSAR) is emerging as a powerful strategy to design drugs with balanced multi-target profiles, particularly relevant in complex diseases such as cancer, neurological disorders, and infectious diseases. Future QSAR approaches will focus on predicting activity spectra rather than activity against a single receptor, aligning with systems pharmacology.[10]
4. Integration with Molecular Docking, Pharmacophore Modeling, and Omics Data
The future of QSAR will not exist in isolation but as part of an integrated computational toolkit. Combining QSAR with molecular docking can provide complementary insights, where docking explores ligand–receptor interactions, and QSAR provides quantitative predictions. Pharmacophore modeling further enhances QSAR by identifying essential structural features for biological activity. Additionally, integration with genomics, proteomics, and metabolomics ("omics" data) will allow QSAR to account for complex biological responses, leading to more realistic and clinically translatable predictions.
5. Future in Precision Medicine
Precision medicine aims to design therapies tailored to individual patients based on genetic, metabolic, and environmental factors. QSAR has the potential to play a central role by predicting drug response variability among different patient populations. For example, patient-specific QSAR models could be developed using pharmacogenomics data to guide personalized therapy. In the future, integrating QSAR with clinical data and biomarker information could accelerate the development of safer and more effective individualized treatments.[9]
CONCLUSION
Quantitative Structure–Activity Relationship (QSAR) has emerged as one of the most powerful and widely used tools in modern drug discovery. From its early foundation in the 1960s with Hansch’s pioneering work to the advanced multidimensional and AI-driven models of today, QSAR has consistently bridged the gap between chemical structure and biological activity. It has provided researchers with predictive insights, enabling the identification, optimization, and screening of drug candidates in a cost-effective and time-efficient manner. QSAR plays a pivotal role not only in accelerating lead optimization and virtual screening but also in toxicity prediction, thereby reducing the reliance on animal testing and minimizing late-stage drug development failures. Its integration with computational chemistry, molecular modeling, and statistical methods has further strengthened its reliability and applicability. Looking forward, QSAR is expected to become a cornerstone of pharmaceutical sciences. The integration of QSAR with artificial intelligence, machine learning, big data analytics, and omics technologies will expand its predictive accuracy and applicability across diverse chemical and biological spaces. Furthermore, the development of multi-target QSAR models will align the technique with the growing emphasis on polypharmacology and personalized medicine. In summary, QSAR continues to evolve as a versatile, predictive, and indispensable tool in drug design. With ongoing advancements, it holds the promise of transforming drug discovery and precision medicine, ensuring the development of safer, more effective, and tailored therapeutics for the future.
REFERENCES

Ubale Asmita, Shingote Vishakha, Quantitative Structure–Activity Relationship (QSAR) in Modern Drug Design: Concepts, Applications, and Future Prospects, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 7, 1778-1794. https://doi.org/10.5281/zenodo.21267104
 10.5281/zenodo.21267104
10.5281/zenodo.21267104