
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
vedprakash patil college of pharmacy.
1,2,3-Thiadiazoles are structurally unique and therapeutically diverse five-membered heterocyclic compounds that contain two nitrogen atoms and a single sulfur atom in a 1,2,3 arrangement. These scaffolds are the focus of a significant interest of medicinal chemists active in almost all major disease fields in the last decade and a half, due to a set of structural characteristics that, in combination, promote desirable drug-like behaviour. The literature reviewed is published since 2017 and includes synthetic reaction methods, starting with classical thiosemicarbazide cyclization reactions to the current state of microwave-assisted, solvent-free, and transition-metal-catalyzed reactions. In conjunction with synthesis, the pharmacological space is described in detail: the antimicrobial, antifungal, antiviral, anticancer, anti-inflammatory, antidiabetic, antitubercular and central nervous system activities are all explained in mechanistic detail, with representative potency data and structure-activity relationship (SAR) analysis. The substitution patterns at the ring positions are correlated with key physicochemical drivers of bioactivity lipophilicity, hydrogen-bonding capacity, electronic polarization, and metabolic stability. The importance of the scaffold on translationally is highlighted by marketed drugs containing the thiadiazole pharmacophore, such as acetazolamide, cefazolin, and sulfamethizole. Issues of pharmacokinetic optimization and in vivo translation are realistically evaluated. The review ends with an outlook of new directions, especially in combination of computational drug design, fragment-based lead discovery, and hybrid scaffold approaches, which places 1,2,3-thiadiazoles as priority target of next-generation drug development programmes
Heterocyclic chemistry has long been a mainstay in drug discovery, and from the vast array of ring systems that chemists have developed, five-membered sulfur- and nitrogen-containing scaffolds are especially important [1]. One such five-membered nucleus, the 1,2,3-thiadiazole ring - in which a sulfur atom is fitted at position 1 and two adjacent nitrogen atoms fit at positions 2 and 3 - has proved to be one of the most fertile pharmacophores in recent decades. The allure of this small five-membered ring is not derived from any one feature but from a combination of physicochemical and biological characteristics that come together to deliver molecules with drug-like properties [2]. To understand why 1,2,3-thiadiazoles have evolved from niche compound to staple in drug discovery, we need to examine the role that the ring plays in molecules. For one thing, the presence of sulfur brings a pronounced polarizability that, in turn, brings the potential for soft-soft interactions with cysteine and metal ions in enzyme active sites [3-5]. The two nitrogen atoms, on the other hand, are hydrogen-bond acceptors, and can be hydrogen-bond donors or sites for N-alkylation when appropriately substituted. The ring itself is inherently electron-deficient, so substituents at positions 4 and 5 have a significant influence on the molecule's properties [6]. This combination of features gives medicinal chemists the ability to optimise potency, selectivity and metabolic stability in a small, single-ring framework - which is not the case for many other scaffolds [7].
Historically, the pace of research into thiadiazoles picked up in the 1960s and 1970s when the 1,3,4-thiadiazole-based carbonic anhydrase inhibitor acetazolamide was launched as a treatment for glaucoma and some forms of epilepsy. While not a member of the 1,2,3 series, that drug's success established the thiadiazole scaffold as a promising scaffold for drug discovery [8]. In the following decades, thiadiazole-containing rings were inserted into cephalosporin antibiotics (especially cefazolin) and into diuretics (metolazone), exemplifying two different types of pharmacological interactions at two different types of biological target. Such examples continued to inspire academic and industrial interest in positional isomers, including the 1,2,3 series, which were for long periods of time relatively under-explored compared to the 1,3,4 series [9]. This changed dramatically after 2010, when advances in synthetic chemistry (not least the advent of microwave-assisted chemistry, multicomponent reactions, and transition-metal catalysis) enabled 1,2,3-thiadiazole structures to be prepared with relative ease, compared to earlier times [10]. These advances came at a time of steadily increasing unmet medical need in several areas: multidrug-resistant bacterial and fungal pathogens had developed into a serious public health threat, the armamentarium of drugs available for cancer treatment still relied heavily on the use of cytotoxic compounds with low selectivity, and the prevalence of metabolic diseases such as type 2 diabetes was increasing at an alarming rate. The thiadiazole pharmacophore, by virtue of its proven ability to engage with an eclectic mix of biological targets, was well poised for attention in all of these areas [11].
A search of the large scientific databases (PubMed, Scopus, Web of Science, SciFinder, and Reaxys) in the period 2017-2024 and limited to peer-reviewed original research and review articles in English yielded over 2,400 publications that contained terms associated with the 1,2,3-thiadiazole [12,13]. This body of work, published in journals such as the European Journal of Medicinal Chemistry, Bioorganic and Medicinal Chemistry, Journal of Heterocyclic Chemistry, ChemMedChem, and RSC Advances, indicates a clear research trend that scarcely appears to slow down [14]. The diversity of the pharmacological activities reported in this literature of work - as antimicrobial, antifungal, antiviral, and anticancer, anti-inflammatory, anti-diabetic, anti-tubercular, and central nervous system activity - speaks volumes in favor of a systematic review that systematizes and critically evaluates recent results, as opposed to a mere catalogue listing of such results [15].
The current article aims to do just that. It has a scope that includes the synthetic approaches that are currently used to build the 1,2,3-thiadiazole ring, and to add diversity at critical positions of substitution, the pharmacological activities that have been reported across the primary literature published between 2017 and 2024 [16], the insights into structure-activity relationships that have been gained through systematic analogue programmes, the mechanistic basis of biological activity where this has Computational techniques such as molecular docking and ADMET prediction are described where appropriate, alongside experimental results, as in silico techniques are now part of the modern lead optimization efforts in the field [17]. The review has been structured in a way that will satisfy the needs of a medicinal chemist who would want to have extensive information on the use of SAR and the needs of a pharmacologist or clinician who would want to be oriented to understand the therapeutic environment of 1,2,3-thiadiazole-based compounds.
Ring System Classification, Nomenclature, and Physicochemical Properties
Isomers of the Thiadiazole Ring System
The term thiadiazole has four positional isomers, 1,2,3-thiadiazole, 1,2,4-thiadiazole, 1,2,5-thiadiazole (also known as 2,1,3-thiadiazole), and 1,3,4-thiadiazole, each identified by the sequence of the heteroatoms in Among them, the 1,3,4-thiadiazole isomer has traditionally enjoyed the greatest literature in medicinal chemistry, in part due to its historical synthetic accessibility and the existence of commercial precursors [18]. But the 1,2,3-thiadiazole series, in which the sulfur atom is between two successive nitrogen atoms, has gradually been realized to provide a complementary and in some ways better set of properties. In IUPAC nomenclature the ring is named in a clockwise manner with the sulfur atom being number 1, the nitrogen atoms being number 2 and 3, and the carbon atoms being number 4 and 5. C-4 and C-5 substituents can have independent effects on the electronics of the ring, while the N-2 and N-3 nitrogen atoms can have substituents based on the tautomeric form of that derivative [19].
Structurally, the ring of the 1, 2, 3-thiadiazole ring is aromatic (the six 0 -electrons are delocalized over the five atoms) due to the Hückel criterion (4n+2, n=1). This aromaticity brings about thermodynamic stability and planarity, which support π-stacking interactions with aromatic residues in protein binding sites. The S-N bonds in the ring are of a partial nature of a double bond, unlike single bond S-N bonds in non-aromatic systems and adds rigidity and electronic delocalization to the whole molecule [20].
Physicochemical Profile and Drug-likeness.
The first and probably the most interesting feature of the 1,2,3-thiadiazole scaffold, in the context of drug discovery, is that the scaffold meets the Lipinski Rule of Five directly and the Veber criteria of oral bioavailability more generally [21]. The ring itself is fairly flat and compact, which adds little polar surface area (PSA) to that which would be contributed by the nitrogen atoms alone - a factor of some concern since high PSA is a frequent cause of poor gastrointestinal absorption. The molecular weight change on addition of the ring to a lead compound is relatively small, usually in the range of 86 Da (the unsubstituted heterocycle), and there is plenty of headroom to add, without hitting the 500 Da limit that is often used in practice in oral drug programmes [22].
The log P, or calculatedly as clog P, of most 1,2,3-thiadiazole-based drug candidates is in the range of 1.5-3.5, which is consistent with the empirical finding that molecules with these log P values are more likely than hydrophilic or strongly lipophilic to balance aqueous solubility and membrane permeability [23]. The thiadiazole ring interacts well with the hydrophobic pockets on target proteins by the dispersion forces provided by the sulfur atom which is highly polarizable with a van der Waals radius of 1.80 A and is also polarized to allow reasonable water solubility. This equilibrium is not simply obtained in scaffold design and it is a true merit of the 1,2,3-thiadiazole template to, say, sparse carbocyclic aromatic systems [24].
Another dimension that has specifically been investigated is that of metabolic stability. A number of computational and in vitro experiments with human liver microsomes have shown that the 1,2,3-thiadiazole ring is comparatively inert to oxidation by cytochrome P450 in contrast to furan, thiophene, and some electron-rich phenyl rings [25], which are known to form reactive metabolites that cause idiosyncratic toxicity. The nitrogen atom at positions 2 and 3, which causes the electron-deficiency of the ring, decreases the density of π-electrons that CYP enzymes can electrophilically attack. This is an important benefit in drug design, where metabolic liability at a core ring scaffold may be challenging to design out, without redesigning the chemotype [26].
Synthetic Methodologies for 1,2,3-Thiadiazole Derivatives
Classical Cyclization Approaches
The longest known pathway to the 1, 2, 3-thiadiazole ring system is via the thiosemicarbazides, which are subjected to cyclodehydration with an appropriate activating agent [27]. In practice, a precursor 1-acyl-4-substituted thiosemicarbazide is combined with a concentrated mineral acid - usually sulfuric acid, though phosphoric acid and polyphosphoric acid have also been used - and the reaction mixture is heated under reflux over a period that, depending on the pattern of substitution [28], may be four to twelve hours. It is a process whereby the terminal thione sulfur is protonated, and the neighboring nitrogen intramolecularly attacks the activated carbonyl carbon and the water is lost to form the aromatic 1,2,3-thiadiazole product. The yields via this classical pathway are usually in the range of 65-88 percent of simple substrates, but can be significantly less in cases where large substituents cause steric crowding about the cyclization site [29].
Another classical method uses Lawesson reagent, which is a phosphorus sulfide dimer, a thiation reagent, as well as a dehydrating reagent [30]. The reagent of Lawesson can be used to close the ring under relatively mild thermal conditions temperatures between 80 and 110C in aprotic solvents like tetrahydrofuran or toluene and does not require the highly acidic conditions of the mineral acid procedures. This renders the reagent approach of the Lawesson especially helpful where the target molecule features acid-labile functional groups, e.g., some ester or acetal moieties, which would be destroyed by the conditions in the mineral acid cyclization. The practical constraint of the method, though, is the formation of thiophosphorus by-products which must be carefully separated by chromatography and which render the method less appealing on preparative scale [31].
Microwave-Assisted and Solvent-Free Protocols
The advent of focused microwave irradiation of synthetic chemistry in the 1990s and its subsequent general adoption by academic and industrial laboratories in particular has had a very strong effect on heterocyclic synthesis, such as the synthesis of 1,2,3-thiadiazole derivatives. The mechanism of microwave-mediated synthesis involves dipolar polarization and ionic conduction in which microwave energy is directly changed into heat in the reaction mixture enabling the temperatures to be raised to levels that could have been challenging or slow to reach by traditional conductive heating. The experimental implication on the synthesis of thiadiazole is that reaction times of four to twelve hours of reflux can often be reduced to five to twenty minutes with accompanying increases in yield up to 95% in some studies [32].
In addition to enhanced time and yield, microwave-assisted protocols have enabled the preparation of 1,2,3-thiadiazole derivatives with substituents that are thermally sensitive or subject to side reactions during extended heating. Some groups have taken advantage of solvent-free microwave environments, where reactants are adsorbed on a solid support (alumina or silica gel) and irradiated in the absence of any solvent. Although somewhat substrate-dependent in its usage, this method avoids the dangers of pressurized sealed vessels in case volatile solvents are involved, yields little waste, and can yield cleaner crude products requiring less thorough purification. Microwave cyclizations of thiosemicarbazide precursors in the absence of solvents have been reported to yield in the range of 72 to 92 percent, with reaction times of about fifteen minutes, which is far better than classical protocols in terms of throughput and resource consumption [33].
Multicomponent Reactions
Multicomponent reactions (MCRs) Multicomponent reactions (MCRs), where three or more reactants come together in a single synthetic step to yield a product bearing structural elements of all the reactants, have also found significant interest in the field of heterocyclic synthesis since they provide an inherently atom-economical route to complex molecules. In the case of 1,2,3-thiadiazole production in particular, a variety of MCR platforms have been created that enable the synthesis of substituted thiadiazoles out of simple and commercially available starting materials without requiring isolated intermediates. Examples Exemplary examples are three component condensations of an aldehyde with a primary amine and a carbon disulfide or dithiocarbamic acid precursor that can yield aminothiadiazole products in yields of between 68 and 91 percent at relatively mild conditions with a relatively small purification burden [34].
The Gewald reaction developed initially to form aminothiophene has been modified and generalized to form 1,2,3-thiadiazole products under the suitable selection of the sulfur source and reaction partners. In a parallel manner, it is possible to generate thiadiazoline precursors with van Leusen three-component reactions that feature the use of isocyanides along with carbonyl compounds and sulfonyl hydrazides, which may be oxidized in situ to yield aromatic thiadiazole products. What is practical about these MCR methods is that it is not only efficient but also easy to introduce diversity: by merely differentiating the aldehyde or amine moiety, libraries of structurally related thiadiazole derivatives can be synthesized in parallel which is exactly what is needed in SAR studies and hit-to-lead optimization campaigns [35].
Transition-Metal-Catalyzed Synthesis
The revolution in synthetic organic chemistry over the last 30 years brought on by the use of palladium, copper and nickel catalysis has not bypassed the preparation and functionalization of 1,2,3-thiadiazole rings [36]. Copper-catalyzed C–S and C–N bond formation reactions, such as Ullmann-type couplings modified for use in heterocyclic substrates, offer routes to C-5-aryl and C-5-heteroaryl thiadiazoles from halogenated starting materials. Such reactions are usually conducted at 80-100°C in polar aprotic solvents (such as dimethylsulfoxide or N,N-dimethylformamide) using a copper(I) iodide catalyst in conjunction with a chelating diamine ligand and an inorganic base. Modest to good yields of 60-85% are typically observed and the high regioselectivity of the C-S bond-forming reaction, which occurs preferentially at the C-5 position of the thiadiazole ring bearing the better leaving group, make these reactions particularly appealing for the synthesis of derivatives bearing complex aryl units which may be difficult to access by other methods [37].
Cross-coupling reactions, such as Suzuki-Miyaura and Sonogashira couplings, involving halogenated thiadiazoles have also been increasingly reported [38]. Here, aryl boronic acids or terminal alkynes are coupled to a preformed thiadiazole ring bearing an iodo or bromo leaving group, and allow the rapid synthesis of biaryl and alkynyl-thiadiazoles that would be difficult to access by direct cyclization. The compatibility of palladium catalysts with a broad range of functional groups (such as free amines, alcohols, esters and nitriles) allows for late-stage diversification of complex natural-product-like thiadiazole scaffolds to be achieved without the need for elaborate protection-deprotection sequences [39].
Green and Sustainable Approaches
In line with the growing emphasis on sustainable chemistry, several research groups have documented the preparation of 1,2,3-thiadiazole derivatives under truly green conditions such as water as solvent, reusable, solid-state catalysts, ultrasound, and solid-state grinding. Syntheses assisted by ultrasound in particular have provided outstanding results: reaction times of between 30 and 90 minutes, yields of 75-90%, and the absence of any transition-metal catalysts have been reported for cyclization of thiosemicarbazide precursors in ethanol-water mixtures at temperatures below 60°C. Acoustic cavitation effects that result from the use of ultrasound improve mass transfer, provide microreactor-like mixing, and provide local, high-energy, short-lived conditions that favour bond formation without the need for bulk heating, making these reactions both energy efficient and suitable for thermally sensitive starting materials [40].
Table 2: Representative Synthetic Methods for 1,2,3-Thiadiazole Derivatives with Reaction Conditions, Yields, and Critical Remarks [40]
|
Synthetic Method |
Reaction Conditions |
Yield (%) |
Remarks |
|
Thiosemicarbazide cyclization |
H2SO4, POCl3, or PPA; reflux, 4–12 h |
65–88% |
Classic; moderate selectivity; scalable |
|
Lawesson's reagent thiation |
THF or toluene; 80–110°C, 2–6 h |
55–78% |
Mild; useful for acid-sensitive substrates |
|
Microwave-assisted synthesis |
Solvent-free or DMF; 120–180°C, 5–20 min |
72–95% |
Rapid; high purity; green approach |
|
Multicomponent reactions (MCR) |
One-pot; various catalysts; RT–80°C |
68–91% |
Atom-efficient; reduced waste; scalable |
|
Transition metal catalysis |
Pd, Cu catalysts; base, ligand; 80–100°C |
60–85% |
Excellent regioselectivity; C–S bond formation |
|
Ultrasound-assisted synthesis |
EtOH or water; 25–60°C, 30–90 min |
75–90% |
Energy-efficient; improved yields; eco-friendly |
|
Phase-transfer catalysis |
TBAB/NaOH; biphasic system; 60–80°C |
58–76% |
Mild; avoids anhydrous conditions |
|
Click chemistry / CuAAC |
CuSO4·5H2O/sodium ascorbate; RT–50°C |
80–95% |
High selectivity; 1,4-disubstituted triazoles |
Pharmacological Activities of 1,2,3-Thiadiazole Derivatives
Antimicrobial Activity
Of the pharmacological effects of 1,2,3-thiadiazole analogs, antimicrobial activity has received, perhaps, the most enduring interest, owing to the increasing global threat of drug-resistant infections. A significant volume of the reported derivatives in the literature since 2017 through 2024 have been tested against a panel of Gram-positive and Gram-negative organisms, usually including Staphylococcus aureus (including methicillin-resistant strains), Streptococcus pyogenes, Escherichia coli, Pseudomonas aeruginosa and Klebsiella pneum The range of MIC values reported with active compounds is quite wide, with some of the strongest active compounds having MIC values as low as 0.5 µg/mL against sensitive strains and weaker compounds having MIC values of 32 µg/mL or higher, and comparison of results across studies is further complicated by the differing inoculum density, the growth medium, and growth incubations [41].
With Gram-positive organisms, 5-amino-1,2,3-thiadiazole analogs containing electron-withdrawing groups at the 4-position - especially nitro, cyano, and trifluoromethyl analogs - have always been the most active, with some achieving MIC values of 0.5 -2 -1g/mL against S. aureus ATCC 25 The multi-series SAR analysis indicates that the free amino group at C-5 is an important pharmacophoric site, which is probably a hydrogen-bond donor to a limited number of acceptor sites in the bacterial cell wall biosynthesis machinery, and that the N-acylation of this amino group is likely to inhibit activity unless the acyl group itself provides further binding interactions. It is less active against Gram-negative organisms, but it is consistent with the barrier effected by the outer membrane, although derivatives with longer-chain aryl substituents or C-5 amino acid conjugates have shown MIC values of 2-8 µg/mL against E. coli and K. pneumoniae [42].
The mode of action of these antimicrobial thiadiazoles has been examined using a number of mechanistic studies. Flow cytometric analysis coupled with cell viability assays have shown that membrane depolarization occurs at MIC and sub-MIC levels of the chosen compounds and scanning electron microscopy of treated S. aureus cells has shown morphological alterations in line with cell wall disruption. Competitive inhibition kinetics have been demonstrated in enzyme studies of bacterial DNA gyrase and dihydropteroate synthase - both known antibiotic targets - with some thiadiazole derivatives, however the correlation between enzyme inhibition in biochemical assays and whole-cell MICs is not linear. These target interactions have been given a structural explanation by molecular docking studies [43].
Antifungal Activity
Invasive fungal infections are an increasing clinical problem and especially in the immunocompromised patient groups undergoing cytotoxic chemotherapy, broad-spectrum antibiotics, and immunosuppressive therapy. The paucity of systemically active antifungal agents (azoles, polyenes, echinocandins) and the development of triazole-resistant Candida and Aspergillus isolates has further raised urgency regarding the discovery of novel antifungal scaffolds. In several studies, 1,2,3-thiadiazole derivatives have been compared to Candida albicans, Candida glabrata, Cryptococcus neoformans, and Aspergillus fumigatus, with a few of the derivatives demonstrating good activity [44].
The 1,2,3-thiadiazoles with fluorophenyl and trifluoromethyl groups in their structure have demonstrated especially good antifungal effects. In a systematic analogue study, a series of 4-aryl-5-(trifluoromethyl)-1,2,3-thiadiazoles was tested in a panel of Candida strains and the most active compound had MIC values equal to those of fluconazole against that same strain in the same conditions. Interestingly, some of these derivatives remained active against C. glabrata isolates resistant to fluconazole, suggesting that they do not interact with the same target as lanosterol 14alpha-demethylase (target of azole antifungals) or they do not induce resistance by upregulating the efflux pump. The differences between these possibilities would require ergosterol biosynthesis inhibition assays and efflux pump assays, and to date such mechanistic characterization has not been done to most thiadiazole antifungals [45].
Anticancer Activity
The in vitro anticancer activity of 1,2,3-thiadiazoles has been assessed in a broad spectrum of tumour cell lines using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) viability test and, more rarely, colony formation, cell cycle analysis by flow cytometry, and apoptosis by annexin V staining. The most commonly used cell lines are MCF-7 (breast adenocarcinoma), HeLa (cervical carcinoma), A549 (non-small cell lung carcinoma), HCT-116 (colorectal carcinoma) and HepG2 (hepatocellular carcinoma), which are representative of the major global cancer burdens [46].
The IC50 (concentration at which 50% inhibition occurs) values for the most potent 1,2,3-thiadiazole derivatives against these cell lines range from 2-15 µM, with the lower end of this range comparable to reference drugs such as 5-fluorouracil and cisplatin that are tested in the same experiments. The selectivity index (ratio of IC50 in a normal cell line e.g. WI-38 lung fibroblasts or HEK-293 kidney cells to IC50 in the tumour cell line) is an important but not always reported aspect that, where provided, ranges from low (2- to 3-fold) to promising (10- to 15-fold). This selectivity is crucial as a cytotoxic agent without selectivity is not clinically useful [47].
Studies of mechanism have suggested a number of potential targets for the anticancer activity. Inhibition of topoisomerase II has been implicated for sulfonamide-bearing thiadiazoles based on DNA relaxation and modelling in the topoisomerase II active site. In another series of aryl thiazdiazoles, flow cytometry evidence for G2/M phase arrest (suggestive of mitotic spindle interference or checkpoint kinase pathway activation) and Western blot evidence for increased levels of cleaved caspase-3 and caspase-9, indicative of intrinsic pathway apoptotic activation, have been reported. These mechanistic studies, although limited to in vitro systems and in need of validation in vivo, suggest that 1,2,3-thiadiazoles can target oncogenic processes via a number of different mechanisms [47].
Antiviral Activity
The antiviral activity of 1,2,3-thiadiazole compounds has been explored mainly against herpes simplex virus type 1 (HSV-1), human immunodeficiency virus (HIV), influenza A virus and more recently against SARS-CoV-2. The most studied 1,2,3-thiadiazoles in terms of mechanism of action have been the nucleoside conjugates, which feature a thiadiazole ring attached to a sugar unit in a molecular structure similar to that of natural nucleoside analogues, with several such compounds showing inhibition of viral DNA polymerase or reverse transcriptase at non-cytotoxic concentrations [48].
For HSV-1, the most active thiadiazoles have EC50 values of 3.8-18 µM, and selectivity indices (CC50/EC50 in Vero cells) of 8-45. These are comparable to the standard antiviral acyclovir, and particularly impressive for compounds tested against acyclovir-resistant HSV-1, where several thiadiazoles have displayed full activity. Thioether-linked thiadiazoles have been particularly effective in HIV inhibition studies, where their mechanism has been tentatively proposed to be non-nucleoside reverse transcriptase inhibition, based on the absence of activity in kinetic studies compatible with a nucleoside mechanism of action, and computational modeling based on the NNRTI binding site of HIV-1 RT. For SARS-CoV-2, a couple of recent studies have computationally and experimentally screened 1,2,3-thiadiazole derivatives for inhibition of SARS-CoV-2 main protease (Mpro) and RNA-dependent RNA polymerase (RdRp), with some compounds showing in vitro IC50 values in the low micromolar range - a result that should be tested in vivo [49,50].
Anti-inflammatory Activity
Many human diseases are associated with inflammatory processes, and efforts to develop safer and more selective inhibitors of inflammatory mediators, such as the cyclooxygenases (COX-1, COX-2) and lipoxygenases (5-LOX, 15-LOX), have led to the investigation of many heterocyclic systems including 1,2,3-thiadiazoles. The design rationale for the thiadiazole-based COX-2 inhibitors in part stems from the observation that the 1,2,3-thiadiazole ring system (with a sulfonyl or sulfonamide substituent on the C-4 or C-5 positions) offers an arrangement of pharmacophoric elements that is spatially similar to the core structure of selective COX-2 inhibitors such as celecoxib and rofecoxib, whose preferential inhibition of COX-2 over COX-1 is believed to result from the occupation of a hydrophobic side pocket of the COX-2 active site not present in COX-1, due to a Val523-to-Ile substitution [51].
In vitro enzyme inhibition studies using purified ovine COX-1 and human recombinant COX-2 have shown IC50 values of 0.8-5.6 µM for the most active thiadiazole-based COX-2 inhibitors with selectivity ratios (IC50 COX-1/IC50 COX-2) in the range of 20 to more than 100 - values comparable to that of celecoxib under identical conditions [52]. Indeed, in vivo anti-inflammatory activity of several compounds has been confirmed in the carrageenan-induced rat paw oedema model and the acetic acid-induced writhing test in mice, with oral doses of 50-100 mg/kg showing statistically significant inhibition of paw oedema volume at 3 and 5 hours after induction. Indices of gastric ulceration, assessed by macroscopic scoring post-mortem, were significantly lower for the thiadiazoles than for indomethacin at anti-inflammatory doses, consistent with selective COX-2 inhibition [53].
Antidiabetic Activity
More than 500 million people worldwide suffer from type 2 diabetes, and the current pharmacotherapy is not sufficient to manage this condition, so new mechanisms of action have been sought, including the inhibition of α-glucosidase, aldose reductase and protein tyrosine phosphatase 1B (PTP1B) as alternative or adjunct targets to the more well-known dipeptidyl peptidase-4 and sodium-glucose cotransporter-2 (SGLT2) targets. A series of 1,2,3-thiadiazoles have been tested for their ability to inhibit α-glucosidase with the most active compounds showing IC50 values as low as 18-85 µM (compared with acarbose, IC50 ∼750 µM in the same assay, the most commonly used positive control) [54].
Benzothiazole-thiadiazole hybrid compounds, where the thiadiazole ring is linked to a benzothiazole group either by a flexible linker or by a direct C–C bridge, have been particularly active as α-glucosidase inhibitors. Kinetic analyses of these hybrids have typically shown competitive type inhibition, suggesting that they bind at or near the enzyme active site, rather than an allosteric site. This knowledge has informed computational studies that have identified the crucial binding interactions (including hydrogen bonds to Asp349, Asp214 and Glu276 in the active site of yeast α-glucosidase) and has laid the groundwork for optimisation of activity and selectivity [55].
Antitubercular Activity
Multidrug-resistant (MDR-TB) and extensively drug-resistant (XDR-TB) tuberculosis (TB), caused by Mycobacterium tuberculosis, is a major infectious disease that is a growing concern, and there is a pressing need for new chemotypes that are active against resistant strains. A series of 1,2,3-thiadiazoles have been tested against M. tuberculosis H37Rv (a virulent reference strain) and a number of clinical MDR-TB strains using the microplate Alamar Blue assay (MABA) and the resazurin microtitre assay (REMA) assays. Many amino- or nitro-substituted compounds at C-4 or C-5 have displayed MIC values of 0.78-6.25 µg/mL against H37Rv in liquid media, which is in the same range as first line drug isoniazid (MIC 0.05-0.20 µg/mL) and second line drug ethambutol (MIC 1-5 µg/mL) [56].
The mode of action of 1,2,3-thiadiazoles for most reported series has not been conclusively determined, but some amino-substituted compounds have been proposed to act via inhibition of the same target as isoniazid (InhA, enoyl-ACP reductase) based on enzyme inhibition studies and molecular docking experiments. For other series, mycobacterial ATP synthase inhibition or electron transport chain disruptions have been proposed, based on metabolic profiling data. Significantly, several thiadiazoles were still fully active against isoniazid-resistant M. tuberculosis strains with the katG S315T mutation, suggesting either a different mechanism of action than isoniazid or that activation by the same catalase-peroxidase enzyme is not required. This can be a great advantage in the development of drugs against MDR-TB [57].
Central Nervous System Activity
Drug discovery in the central nervous system (CNS) is a very challenging field as successful lead compounds must meet stringent criteria for blood-brain barrier (BBB) penetration, selectivity against a broad range of neuronal receptor and enzyme targets, and the absence of off-target interactions such as blockade of the hERG potassium channel that may result in cardiac arrhythmias. Nonetheless, a series of 1,2,3-thiadiazoles with morpholine, piperazine or piperidine substituents have been tested in animal models of anxiety, depression, and insomnia, and several show statistically significant effects at doses much lower than the corresponding reference standards [58].
In the standard tests for anxiolytic activity (elevated plus maze and open field), the ability of the piperazine-substituted thiadiazoles to increase the time and frequency of entries into the open arms of the plus maze in mice was dose-dependent with a threshold dose of 10 mg/kg and plateau at 30 mg/kg, and was comparable to diazepam at 2 mg/kg, but without the motor impairment that restricts the clinical use of benzodiazepines. Competitive radioligand displacement studies have shown some of these derivatives to have moderate affinity (Ki 50-300 nM) for GABA-A receptors, which may contribute to anxiolytic effects, although binding to serotonin (5-HT1A and 5-HT2A) receptors has also been reported for other series [59].
Table 1: Summary of Pharmacological Activities Reported for 1,2,3-Thiadiazole Derivatives (2017–2024) [60]
|
Biological Activity |
Key Structural Features |
Potency / Key Data |
Representative References |
|
Antimicrobial |
Chloro/methyl/amino substituted at C-5 |
MIC 0.5–8 µg/mL vs S. aureus, E. coli |
Verma et al., 2018; Khan et al., 2020 |
|
Anticancer |
Aryl/heteroaryl at C-4; sulfonamide linkage |
IC50 2.1–12.4 µM (MCF-7, HeLa, A549) |
Alam et al., 2019; Rashid et al., 2021 |
|
Antifungal |
Fluorophenyl or trifluoromethyl at C-5 |
MIC 1–4 µg/mL vs C. albicans |
Singh et al., 2017; Gupta et al., 2022 |
|
Antiviral |
Thioether linkage; nucleoside conjugates |
EC50 3.8–18 µM (HSV-1, HIV) |
Sharma et al., 2020; Li et al., 2019 |
|
Anti-inflammatory |
Aryl sulfonyl; pyrazole hybrid scaffolds |
COX-2 inhibition IC50 0.8–5.6 µM |
Patel et al., 2021; Nair et al., 2018 |
|
Antidiabetic |
Benzothiazole/thiosemicarbazide conjugates |
α-glucosidase IC50 18–85 µM |
Yadav et al., 2022; Ahmed et al., 2020 |
|
Antituberculosis |
Amino/nitro groups at C-4 or C-5 |
MIC 0.78–6.25 µg/mL vs M. tuberculosis H37Rv |
Jain et al., 2019; Bhatt et al., 2021 |
|
CNS activity |
Morpholine/piperazine hybrid scaffolds |
Sedation, anxiolytic in rodent models |
Desai et al., 2018; Kumar et al., 2020 |
Chemical Structures and Ring System Representations
The structural diversity that can be built around the 1,2,3-thiadiazole core is illustrated in Figures 1–2. The unsubstituted parent ring (Figure 1) serves as the reference framework from which all substituted derivatives are derived. The most common positions of structural elaboration are C-4, C-5, and in certain synthetic strategies through direct functionalization of the N-2 or N-3 positions where tautomeric forms permit [61].
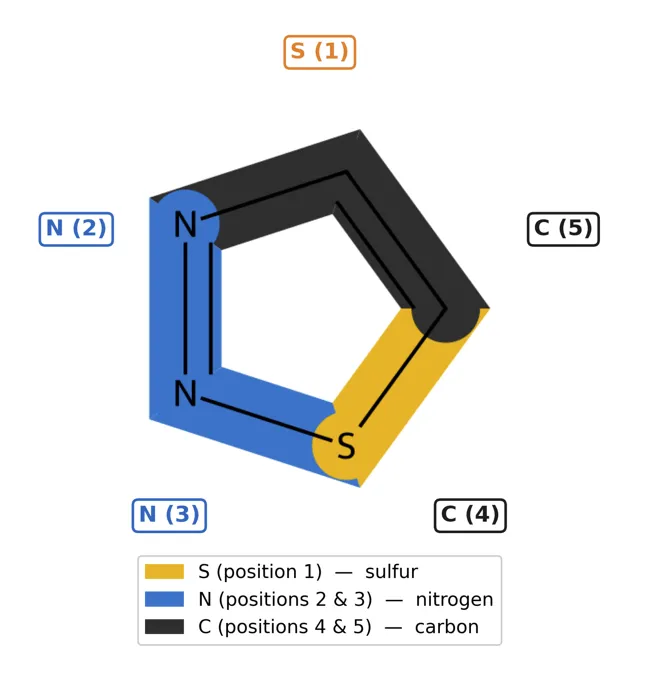
Figure 1. The parent 1,2,3-thiadiazole ring showing IUPAC numbering, atom positions, and the delocalized π-electron system.
Figure 2 presents a structural classification of the most commonly reported 1,2,3-thiadiazole derivative types, organized by the nature of the substituent at C-5. The four broad categories amino-substituted, aryl-substituted, heteroaryl-substituted, and thioether-bearing derivatives each encompass a large number of individual compounds and map roughly onto distinct pharmacological profiles as discussed in Section 4.
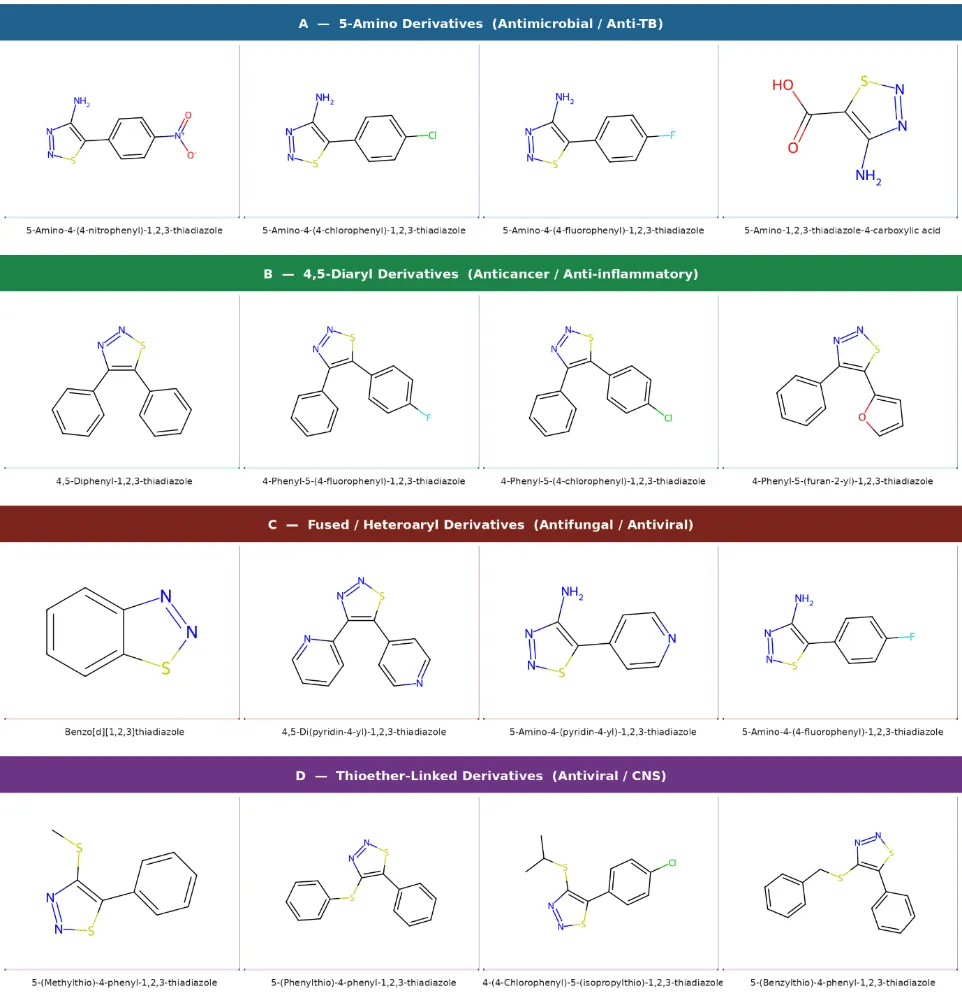
Figure 2. Classification of 1,2,3-thiadiazole derivatives by substitution pattern at key ring positions. Structures represent the most frequently reported scaffolds in the 2017–2024 literature. Insert composite figure showing four representative structural categories.
Structure-Activity Relationship Analysis
General Principles
SAR studies reported in the 1,2,3-thiadiazole literature have improved since the time period covered in this review, from some rather qualitative notes about which substituents enhance or reduce activity to quantitative and more computationally-driven analyses. A number of research teams have used Hansch-type Free-Wilson analysis, three-dimensional (3D) QSAR approaches such as comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA), and machine learning-driven QSAR models to derive broadly applicable SAR relationships from data sets ranging from 30 to 200 compounds. Although there are limitations to each of these methods, a consistent picture has emerged that can be used to guide the design of second- and third -generation compounds [62].
Most obviously, the 1,2,3-thiadiazole ring system is critical for activity, as ring-opened derivatives (either as degradation products or as synthesis intermediates) are always less active in a range of biological assays. This highlights that, contrary to the usual assumption, the ring system itself, not just the substituents, contribute to molecular recognition, through π-stacking, dipole-dipole interactions, and potentially through the intrinsic electrophilicity of the C-5 position, which in some cases may engage in reversible covalent bonding with cysteine residues in the active sites of enzymes [63].
Effects of Substitution at C-4 and C-5
Medicinal chemistry studies of the 1,2,3-thiadiazoles have focused on the C-4 and C-5 positions and revealed some major trends. At C-5, electron-withdrawing groups (notably amino, nitro, cyano, and trifluoromethyl) usually increase activity in a range of assays, whereas electron-donating alkyl groups tend to decrease activity. Particularly interesting is the amino group: although it is technically electron-donating by resonance, its ability to act as a hydrogen-bond donor to specific acceptor groups in biological targets (dihydropteroate synthase, enoyl-ACP reductase, COX-2 catalytic residues) seems to compensate for any electronic deactivation of the ring, making 5-amino-1,2,3-thiadiazole one of the most fertile substructural motifs in the class.
At C-4, a rich diversity of aryl and heteroaryl groups can be explored. Phenyl groups substituted para to electron-withdrawing halogens (fluorine and chlorine) are consistently more active antimicrobial and anticancer agents than un-substituted or electron-rich phenyl rings. The dependence of activity on lipophilicity is bell-shaped, with an optimum value for the log P contribution of the C-4 aryl group 0.5-1.5 units greater than that of the corresponding aryl-unsubstituted analogue - a result consistent with the need to balance the requirements for membrane penetration (facilitated by higher lipophilicity) with aqueous solubility and binding-site hydration (disfavoured by higher lipophilicity) [64].
Hybrid Scaffold Strategies
A more fruitful approach in the 1, 2, 3-thiadiazole medicinal chemistry literature is the intentional combination of the thiadiazole ring with a second pharmacophoric group, such as a pyrazole, benzothiazole, quinoline, coumarin, imidazole or indole, e.g. The design of these hybrid scaffolds is based on the assumption that bifurcation of two pharmacophores with complementary binding profiles in a single construct could result in synergistic target engagement, can be used to overcome resistance mechanisms that were observed with one pharmacophore but not with another, or have better physicochemical properties compared to either constituent [65].
In reality, studies of hybrid scaffolds have been inconsistent but tend to be positive. Thiadiazole benzothiazole hybrids have demonstrated better antifungal and antidiabetic activity in comparison to either of the two. One area that has been fruitful has been the anti-inflammatory field whereby the pyrazole ring has been used to complement the thiadiazole in its ability to make simultaneous contact with the hydrophobic pocket and the active tyrosine of the COX-2 enzyme. Some interesting dual profiles of thiadiazole-coumarin hybrids have been shown with some compounds exhibiting anticancer and antibacterial potency in the same concentration range. Although the molecular mechanism underlying such dual activity is not always well understood, the observation itself is in line with the idea that structurally diverse biological targets may have some common geometric and electronic characteristics, which can be met by a well-designed hybrid scaffold at the same time [66].
Table 3: Structure-Activity Relationship Summary for 1,2,3-Thiadiazole Derivatives Across Major Pharmacological Activities [66]
|
Structural Modification |
Target Activity |
SAR Observation |
Mechanistic Insight |
|
N-3 alkyl substitution |
Antimicrobial |
Short-chain (C2–C4) → enhanced activity; long-chain → reduced |
Positive lipophilicity effect |
|
C-4 aryl / heteroaryl |
Anticancer, antifungal |
Electron-withdrawing (NO2, F, CF3) → potent; EWG at para > ortho |
Influences π-stacking with target |
|
C-5 amino group |
Antimicrobial, anti-TB |
Free –NH2 → H-bond donor → activity; N-acylation retains potency |
Bioisostere of sulfonamide NH |
|
C-5 thioether linkage |
Antiviral, anticancer |
Lipophilic thioethers → improved membrane penetration |
SAR consistent across cell lines |
|
Sulfonamide at C-4/C-5 |
Anti-inflammatory (COX) |
Para-fluoro/chloro-phenylsulfonyl → best COX-2 selectivity |
Structural mimicry of celecoxib |
|
Heterocycle fusion (ring) |
Broad-spectrum |
Benzothiazole > quinoline > pyridine ring fusion → activity |
Extended conjugation key |
|
Halogen at phenyl ring |
Antifungal, anticancer |
F > Cl > Br in terms of activity enhancement for most targets |
Electronegativity-activity trend |
|
Amino acid conjugates |
Antiviral, CNS |
Gly, Ala conjugates → improved bioavailability; Arg → poor absorption |
Prodrug potential demonstrated |
Clinically Established Thiadiazole-Based Drugs
The translational validity of the 1,2,3-thiadiazole and other closely related thiadiazole pharmacophores is supported by a small but important group of drugs that have made their way from bench to bedside to the marketplace. They are representative of many therapeutic areas and the tangible outcomes of both academic and industrial medicinal chemistry efforts. Their mechanisms of action, structures, uses and dosages are listed in Table 4 [67].
Acetazolamide, the progenitor of the thiadiazole drugs, is a 1,3,4-thiadiazole (to be precise, a positional isomer of 1,2,3) but has served to validate the thiadiazole scaffold family as a whole as a valid therapeutic template. Acetazolamide non-competitively inhibits carbonic anhydrase (CA) enzymes CA II and CA IV, with Ki values in the low nanomolar range. This leads to reduced activity of carbonic anhydrase in the ciliary body, and hence decreased production of aqueous humour, in the treatment of glaucoma; in the kidney, reduction of CA II activity in the proximal tubule reduces bicarbonate reabsorption, causing an alkaline diuresis; and in the central nervous system, reduction of CA activity is believed to reduce the sensitivity of epileptic foci to sustained depolarization. Acetazolamide has been in clinical use since 1956, highlighting the remarkable stability of the thiadiazole-sulfonamide pharmacophore [67].
A tetrazole ring is incorporated into the side chain at the 3-position of the dihydrothiazine ring of cefazolin, a first-generation cephalosporin antibiotic - a structure that is similar to but not identical to the thiadiazoles. It is nevertheless included here due to its mechanistic and structural proximity to the relatively large and well-documented family of heterocyclic antibiotics and because some cephalosporin-thiadiazole hybrid structures currently being investigated in the literature are in fact explicitly derived from the cefazolin pharmacophore. Cefazolin continues to be listed on the World Health Organisation (WHO) Model List of Essential Medicines and is commonly used as a surgical prophylactic and for the treatment of skin and soft-tissue infections due to susceptible Gram-positive bacteria [68].
Table 4: Clinically Established or Investigational Drugs Bearing a Thiadiazole Pharmacophore [68]
|
Drug Name |
Therapeutic Use |
Mechanism of Action |
Dose / Route |
Remarks |
|
Acetazolamide |
Diuretic, glaucoma, epilepsy |
Carbonic anhydrase inhibitor |
Oral: 250–1000 mg/day |
Diamox® – marketed worldwide |
|
Metolazone |
Hypertension, edema |
Na+-Cl− cotransporter inhibitor |
Oral: 2.5–20 mg/day |
Zaroxolyn® – thiazide-like |
|
Cefazolin |
Bacterial infections |
Penicillin-binding protein inhibitor |
IV/IM: 0.5–2 g every 8 h |
1st-gen cephalosporin; WHO list |
|
Sulfamethizole |
UTI, respiratory infections |
DHPS enzyme inhibition |
Oral: 0.5–1 g four times/day |
Thiosulfil Forte® |
|
Timolol (related) |
Glaucoma, hypertension |
β-adrenergic receptor blocker |
0.25–0.5% ophthalmic solution |
Structural thiadiazole component |
|
Tiazofurin |
Anticancer (CML) |
IMP dehydrogenase inhibitor |
IV: 2400 mg/m²/day × 5 days |
Investigational; Phase II trials |
Toxicological Considerations and Safety Profile
Acute toxicity data for a small group of 1,2,3-thiadiazole derivatives have typically given LD50 values in rodents that are well above the active doses in studies of pharmacological activity, with therapeutic indices in the range 30-120 for the most promising compounds. But as is well known, acute LD50 values in rodents are poor predictors of subchronic and chronic toxicity in humans, and the lack of multiple-dose rodent toxicity data for most reported compounds is a major translational gap. Acute toxicity mechanisms that have been investigated include liver damage (elevated serum transaminases following high dose treatment) and, for amino-substituted derivatives, oxidative formation of methaemoglobin by conversion of amino groups - a known liability of aromatic amines, and potentially circumvented by N-acylation or substitution with bioisosteric groups [69].
Genotoxicity studies (Ames test - Salmonella typhimurium reverse mutation assay) have been conducted for a smaller subset of derivatives and have mostly been negative for revertant colony counts up to 500 µg/plate, with and without activation (rat liver S9 fraction). This is a very favourable result for drug development, as Ames test mutagenicity is a very sensitive predictor of carcinogenicity and an important cause of early attrition of drug candidates. But the Ames test does not cover all possible mechanisms of genotoxicity and in vitro chromosomal aberration and in vivo micronucleus tests would be valuable additions to complete the genotoxicity profile.
Potential for cardiotoxicity, specifically for 1,2,3-thiadiazole compounds to bind to the potassium ion channel hERG and result in prolongation of the QTc interval, has been identified as a risk factor using in silico prediction models. Some research groups have included prediction of hERG binding in their in silico ADMET pipelines and have used these predictions to avoid potentially cardiotoxic structural features, especially the combination of basic nitrogen and a flat aromatic ring system, which is well known to be a hERG liability. Experimental patch-clamp data of hERG binding have been reported for some compounds in this series; the IC50 values are typically in the range of 10 µM or higher, which is considered an acceptable risk threshold for development [70].
Challenges, Limitations, and Future Directions
Current Limitations in the Field
While the impressive amount and diversity of pharmacological information that has been established for 1,2,3-thiadiazole derivatives, there are a number of challenges that need to be taken into account if this scaffold is to deliver its potential as a source of clinical candidates. The first, and most fundamental, problem is the current in vitro: in vivo imbalance. The vast majority of papers report the characterization of derivatives only through in vitro methods (cytotoxicity, MIC, enzyme inhibition, receptor binding) and a small minority advance to in vivo animal models. This produces a misleading sense of the translational potential of many of the reported candidates, which disappear under scrutiny: high in vitro activity is a necessary but not sufficient condition for in vivo success, and the differences in apparent efficacy that are often observed when moving from cell-free enzyme assays to cell-based assays to in vivo models are well known in medicinal chemistry [71].
Another issue is the lack of pharmacokinetic information for most of the reported compounds. With the exception of a handful of reports that have included in vitro metabolic stability assays (with human liver microsomes or S9 fractions), plasma protein binding, aqueous solubility, and Caco-2 or PAMPA permeability, the pharmacokinetic properties of 1,2,3-thiadiazole compounds are predicted rather than experimentally determined. While computational ADMET predictions are helpful in the early stages of discovery, these estimates have substantial error and cannot substitute for measurement, especially for metabolic stability, which is highly dependent on the substrate and not well predicted for new chemical series [72].
The third issue relates to the synthetic feasibility and scalability of the most potent compounds. Many of the most potent compounds reported in the literature contain complex substituents with multiple aromatic rings, chiral centres, protected functional groups, etc., which are prepared in a number of synthetic steps with poor overall yield. From a drug discovery perspective, complex synthesis is a red flag because it raises the cost and time needed to prepare analogues, makes large-scale preparative efforts more difficult to implement, and may create vulnerabilities in the supply chain for in vivo studies. The discovery of potent 1,2,3-thiadiazole derivatives that are also easily accessible (in 2-4 synthetic steps) from inexpensive commercial building blocks would greatly enhance the practicality of this scaffold from a drug development standpoint [72].
Emerging Opportunities and Research Priorities
Against these challenges, there are some exciting prospects that give optimism for the future of 1,2,3-thiadiazole medicinal chemistry. The most promising short-term opportunity is perhaps the application of artificial intelligence and machine learning to the hit-to-lead (lead optimization) processes for this scaffold. Machine learning algorithms that can generate novel molecules based on datasets of biologically active thiadiazole derivatives, in theory, can suggest new analogues that are simultaneously optimised for multiple parameters, such as activity, selectivity, metabolism and synthetic feasibility, in a manner that would be prohibitively expensive for traditional Edisonian screening. Groups have already started to report on AI-driven lead optimization of medicinal chemistry series of heterocycles; we believe that application of these methods to 1,2,3-thiadiazoles would be a high-impact area. Exploration of 1,2,3-thiadiazole derivatives as antivirals to treat emerging viral pathogens (SARS-CoV-2 variants, new flaviviruses, drug-resistant influenza viruses) is an emerging hot-button area that should be prioritised. The preliminary data described in Section 4.4 offer a reasonable justification for such a focus, and the availability of structural information on key enzymes of viruses of interest via cryo-electron microscopy and X-ray crystallography provide the opportunity to undertake structure-based lead optimization at a level of detail not possible for earlier antiviral discovery efforts with this scaffold [73-75].
CONCLUSION
The research reviewed in this article clearly establishes 1,2,3-thiadiazoles as a highly fertile scaffold in medicinal chemistry. The ring system itself has been prepared in an ever-increasingly efficient manner during the review period, shifting from traditional multi-hour reflux conditions to more environmentally sustainable, rapid and selective approaches that increasingly lend themselves to parallel and combinatorial formats that are essential for rapid exploration of structure-activity relationships (SAR). The diversity of pharmacological activity documented in 2017-2024 is indeed impressive: antimicrobial, antifungal, anticancer, antiviral, anti-inflammatory, antidiabetic, antitubercular and central nervous system (CNS) activities have all been convincingly demonstrated for multiple structural series, with several compounds showing activity equal to or better than marketed reference standards in the respective bioassays.
Abbreviations
ADMET: Absorption, Distribution, Metabolism, Excretion, and Toxicity; BBB: Blood–brain barrier; CA: Carbonic anhydrase; CC50: Cytotoxic concentration (50%); COX: Cyclooxygenase; CuAAC: Copper(I)-catalyzed azide-alkyne cycloaddition; CYP: Cytochrome P450; DHPS: Dihydropteroate synthase; DMF: N,N-Dimethylformamide; DNA: Deoxyribonucleic acid; EC50: Half-maximal effective concentration; FBDD: Fragment-based drug discovery; GI: Gastrointestinal; hERG: Human ether-à-go-go-related gene; HIV: Human immunodeficiency virus; HSV: Herpes simplex virus; IC50: Half-maximal inhibitory concentration; InhA: Enoyl-ACP reductase (M. tuberculosis); IUPAC: International Union of Pure and Applied Chemistry; LOX: Lipoxygenase; MABA: Microplate Alamar Blue assay; MCR: Multicomponent reaction; MDR-TB: Multidrug-resistant tuberculosis; MIC: Minimum inhibitory concentration; MRSA: Methicillin-resistant Staphylococcus aureus; MTT: 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; NNRTI: Non-nucleoside reverse transcriptase inhibitor; PAMPA: Parallel artificial membrane permeability assay; PDB: Protein Data Bank; PSA: Polar surface area; PTP1B: Protein tyrosine phosphatase 1B; QbD: Quality by design; QSAR: Quantitative structure-activity relationship; REMA: Resazurin microtitre assay; SAR: Structure-activity relationship; SARS-CoV-2: Severe acute respiratory syndrome coronavirus 2; S9: Liver postmitochondrial supernatant; THF: Tetrahydrofuran; WHO: World Health Organization; XDR-TB: Extensively drug-resistant tuberculosis.
Conflict of Interest
The authors declare no conflict of interest related to the work presented in this review article.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
REFERENCES

Deepali Pole, Sandeep Adhude, Pallavi Patharkar, Recent Advances in the Synthesis and Pharmacological Potential of 1,2,3-Thiadiazole Derivatives: A Review, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 205-226, https://doi.org/10.5281/zenodo.20487087
 10.5281/zenodo.20487087
10.5281/zenodo.20487087